



1. ຄວາມເຂົ້າໃຈເບື້ອງຕົ້ນ
ໃນຂັ້ນຕອນນີ້, ພວກເຮົາຈໍາເປັນຕ້ອງເຂົ້າໃຈບາງແນວຄວາມຄິດແລະຄໍາສັບຕ່າງໆ, ເພື່ອຫຼີກເວັ້ນການເຮັດຜິດພາດຕໍ່ຫນ້າຜູ້ສູງອາຍຸຂອງພວກເຮົາ, ເຊັ່ນ:
Q: ຄວາມແຕກຕ່າງກັນລະຫວ່າງ RT-PCR, qPCR, Real-time PCR ແລະ RT-PCR ໃນເວລາຈິງແມ່ນຫຍັງ?
ຄໍາຕອບ: RT-PCR ແມ່ນ PCR transcription reverse(PCR ການຖອດຂໍ້ຄວາມແບບປີ້ນກັບກັນ, RT-PCR), ເຊິ່ງເປັນຕົວແປທີ່ໃຊ້ກັນຢ່າງກວ້າງຂວາງຂອງປະຕິກິລິຍາລະບົບຕ່ອງໂສ້ polymerase (PCR).ໃນ RT-PCR, strand RNA ຖືກຖອດຖອນຄືນເປັນ DNA ເສີມ, ເຊິ່ງຫຼັງຈາກນັ້ນຖືກນໍາໃຊ້ເປັນແມ່ແບບສໍາລັບການຂະຫຍາຍ DNA ໂດຍ PCR.
ເວລາຈິງ-PCR ແລະ qPCR(Quantitative Rea-ltime-PCR) ແມ່ນສິ່ງດຽວກັນ, ທັງສອງແມ່ນ PCR ປະລິມານທີ່ແທ້ຈິງ, ຊຶ່ງຫມາຍຄວາມວ່າແຕ່ລະວົງຈອນຂອງ PCR ມີບັນທຶກຂໍ້ມູນໃນເວລາທີ່ແທ້ຈິງ, ດັ່ງນັ້ນຈໍານວນຂອງແມ່ແບບເລີ່ມຕົ້ນສາມາດປັບການວິເຄາະທີ່ຊັດເຈນ.
ເຖິງແມ່ນວ່າທັງສອງ PCR ເວລາຈິງ (PCR ປະລິມານ fluorescent ທີ່ແທ້ຈິງ) ແລະ PCR transcription PCR (ການຖອດຂໍ້ຄວາມແບບປີ້ນກັບ PCR) ເບິ່ງຄືວ່າຖືກຫຍໍ້ເປັນ RT-PCR, ສົນທິສັນຍາສາກົນແມ່ນ: RT-PCR ໂດຍສະເພາະຫມາຍເຖິງການຖອດຂໍ້ຄວາມແບບປີ້ນກັບກັນ.PCR , PCR ໃນເວລາຈິງໂດຍທົ່ວໄປແມ່ນຫຍໍ້ເປັນ qPCR (PCR ໃນເວລາຈິງປະລິມານ).
ແລະເວລາຈິງ RT-PCR (RT-qPCR), ມັນແມ່ນ PCR ການຖອດຂໍ້ຄວາມແບບປີ້ນກັບກັນລວມກັບເທກໂນໂລຍີປະລິມານ fluorescent: ທໍາອິດໄດ້ຮັບ cDNA (RT) ຈາກ RNA reverse transcription, ແລະຫຼັງຈາກນັ້ນໃຊ້ PCR ເວລາຈິງສໍາລັບການວິເຄາະປະລິມານ (qPCR).ຫ້ອງທົດລອງສ່ວນໃຫຍ່ເຮັດ RT-qPCR, ນັ້ນແມ່ນ, ການຄົ້ນຄວ້າກ່ຽວກັບ RNA expression down-regulation, ດັ່ງນັ້ນ qPCR ທີ່ທຸກຄົນເວົ້າກ່ຽວກັບຫ້ອງທົດລອງຕົວຈິງຫມາຍເຖິງ RT-qPCR, ແຕ່ຢ່າລືມວ່າຍັງມີການທົດສອບ DNA ຫຼາຍຢ່າງໃນການນໍາໃຊ້ທາງດ້ານການຊ່ວຍ.ການວິເຄາະດ້ານປະລິມານ, ເຊັ່ນ: ການກວດຫາເຊື້ອໄວຣັສຕັບອັກເສບ B.
ຄໍາຖາມ: ຫຼັງຈາກອ່ານ PCR ປະລິມານ fluorescent ຫຼາຍ, ເປັນຫຍັງຊິ້ນສ່ວນຂະຫຍາຍໃຫຍ່ຄວນຖືກຄວບຄຸມພາຍໃນຂອບເຂດຂອງ 80-300bp?
ຄໍາຕອບ: ຄວາມຍາວຂອງແຕ່ລະ gene sequence ແຕກຕ່າງກັນ, ບາງອັນແມ່ນຫຼາຍ kb, ບາງອັນແມ່ນຫຼາຍຮ້ອຍ bp, ແຕ່ພວກເຮົາພຽງແຕ່ຕ້ອງການໃຫ້ຄວາມຍາວຂອງຜະລິດຕະພັນແມ່ນ 80-300bp ເມື່ອອອກແບບ primers, ສັ້ນເກີນໄປຫຼືຍາວເກີນໄປແມ່ນບໍ່ເຫມາະສົມສໍາລັບການກວດສອບ PCR ປະລິມານ fluorescent.ຊິ້ນສ່ວນຂອງຜະລິດຕະພັນແມ່ນສັ້ນເກີນໄປທີ່ຈະແຍກອອກຈາກ primer-dimer.ຄວາມຍາວຂອງ primer-dimer ແມ່ນປະມານ 30-40bp, ແລະມັນຍາກທີ່ຈະຈໍາແນກວ່າມັນເປັນ primer-dimer ຫຼືຜະລິດຕະພັນຖ້າມັນຫນ້ອຍກວ່າ 80bp.ຖ້າຊິ້ນສ່ວນຜະລິດຕະພັນຍາວເກີນໄປ, ເກີນ 300bp, ມັນຈະເຮັດໃຫ້ເກີດປະສິດທິພາບການຂະຫຍາຍຕໍ່າແລະບໍ່ສາມາດກວດພົບປະລິມານຂອງ gene ໄດ້ຢ່າງມີປະສິດທິພາບ.
ຕົວຢ່າງ, ເມື່ອທ່ານນັບຈໍານວນຄົນຢູ່ໃນຫ້ອງຮຽນ, ທ່ານພຽງແຕ່ຕ້ອງນັບຈໍານວນປາກເທົ່ານັ້ນ.ອັນດຽວກັນແມ່ນເປັນຄວາມຈິງໃນເວລາທີ່ທ່ານກວດພົບ gene, ທ່ານພຽງແຕ່ຕ້ອງການກວດສອບລໍາດັບສະເພາະໃດຫນຶ່ງຂອງ gene ເພື່ອເປັນຕົວແທນຂອງລໍາດັບທັງຫມົດຈະເຮັດໄດ້.ຖ້າຢາກນັບຄົນກໍ່ຕ້ອງນັບທັງປາກ ແລະດັງ, ຫູ, ແວ່ນ, ຈື່ງເຮັດຜິດງ່າຍ.
ເພື່ອຂະຫຍາຍ, ໃນການຄົ້ນຄວ້າທາງຊີວະວິທະຍາ, ມີຫຼາຍກໍລະນີຄົ້ນຄ້ວາຈາກຈຸດໄປຫາພື້ນທີ່, ເນື່ອງຈາກວ່າລໍາດັບ gene ຂອງຊະນິດພັນໃດແມ່ນຍາວຫຼາຍ, ມັນບໍ່ຈໍາເປັນແລະເປັນໄປບໍ່ໄດ້ທີ່ຈະວັດແທກ fragments ທັງຫມົດ, ເຊັ່ນ: ລໍາດັບເຊື້ອແບັກທີເຣັຍ 16S, ເຊິ່ງເປັນການປະຕິບັດລໍາດັບການອະນຸລັກຂອງເຊື້ອແບັກທີເຣັຍ Assays ເພື່ອ infer ຈໍານວນປະຊາກອນທີ່ແນ່ນອນຂອງເຊື້ອແບັກທີເຣັຍ.
Q: ຄວາມຍາວທີ່ດີທີ່ສຸດສໍາລັບການອອກແບບ primer qPCR ແມ່ນຫຍັງ?
ຄໍາຕອບ: ໂດຍທົ່ວໄປແລ້ວ, ຄວາມຍາວ primer ແມ່ນປະມານ 20-24bp, ເຊິ່ງດີກວ່າ.ແນ່ນອນ, ພວກເຮົາຕ້ອງເອົາໃຈໃສ່ກັບຄ່າ TM ຂອງ primer ເມື່ອອອກແບບ primer, ເພາະວ່ານີ້ແມ່ນກ່ຽວຂ້ອງກັບອຸນຫະພູມທີ່ດີທີ່ສຸດ.ຫຼັງຈາກການທົດລອງຫຼາຍ, ມັນໄດ້ຖືກພິສູດວ່າ 60 ° C ແມ່ນຄ່າ TM ທີ່ດີກວ່າ.ຖ້າອຸນຫະພູມ annealing ຕ່ໍາເກີນໄປ, ມັນໄດ້ຢ່າງງ່າຍດາຍຈະນໍາໄປສູ່ການຂະຫຍາຍທີ່ບໍ່ສະເພາະ.ຖ້າອຸນຫະພູມ annealing ສູງເກີນໄປ, ປະສິດທິພາບການຂະຫຍາຍຈະຂ້ອນຂ້າງຕ່ໍາ, ສູງສຸດຂອງເສັ້ນໂຄ້ງຂອງ amplification ຈະເລີ່ມຕົ້ນຕໍ່ມາ, ແລະຄ່າ CT ຈະຊັກຊ້າ.
ຖາມ: ວິທີການຍ້ອມສີແຕກຕ່າງຈາກວິທີການກວດສອບແນວໃດ?
ຄໍາຕອບ: ວິທີການຍ້ອມສີຍ້ອມ fluorescent ບາງອັນ, ເຊັ່ນ: SYBR Green Ⅰ, PicoGreen, BEBO, ແລະອື່ນໆ, ບໍ່ປ່ອຍແສງດ້ວຍຕົວມັນເອງ, ແຕ່ຈະປ່ອຍ fluorescence ຫຼັງຈາກຜູກມັດກັບຮ່ອງເລັກນ້ອຍຂອງ DNA ສອງສາຍ.ດັ່ງນັ້ນ, ໃນຕອນເລີ່ມຕົ້ນຂອງປະຕິກິລິຍາ PCR, ເຄື່ອງບໍ່ສາມາດກວດພົບສັນຍານ fluorescent ໄດ້.ເມື່ອປະຕິກິລິຍາມາຮອດຂັ້ນຕອນການຂະຫຍາຍການເນລະມິດ, ສາຍສອງຖືກເປີດ, ແລະສາຍພັນໃໝ່ຖືກສັງເຄາະພາຍໃຕ້ການກະທຳຂອງ DNA polymerase, ແລະໂມເລກຸນ fluorescent ຜູກມັດກັບ dsDNA ຮ່ອງເລັກນ້ອຍ.ເມື່ອຈໍານວນຂອງວົງຈອນ PCR ເພີ່ມຂຶ້ນ, ສີຍ້ອມຫຼາຍແລະຫຼາຍໄດ້ຖືກລວມເຂົ້າກັບ DNA ຄູ່, ແລະສັນຍານ fluorescent ຍັງໄດ້ຮັບການປັບປຸງຢ່າງຕໍ່ເນື່ອງ.ວິທີການຍ້ອມສີແມ່ນຖືກນໍາໃຊ້ຕົ້ນຕໍໃນການຄົ້ນຄວ້າວິທະຍາສາດ.
PS: ລະມັດລະວັງໃນເວລາເຮັດການທົດລອງ, ສີຍ້ອມຕ້ອງປະສົມກັບ DNA ຂອງມະນຸດ, ລະມັດລະວັງທີ່ຈະເຮັດໃຫ້ມັນກາຍເປັນ fluorescent.
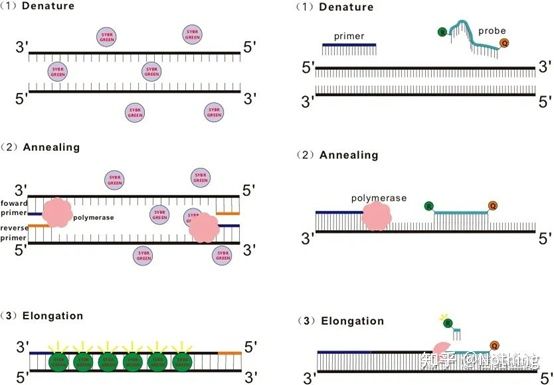
ວິທີການຍ້ອມສີ (ຊ້າຍ) ວິທີການ Probe (ຂວາ)
PS: ລະມັດລະວັງໃນເວລາເຮັດການທົດລອງ, ສີຍ້ອມຕ້ອງປະສົມກັບ DNA ຂອງມະນຸດ, ລະມັດລະວັງທີ່ຈະເຮັດໃຫ້ມັນກາຍເປັນ fluorescent.
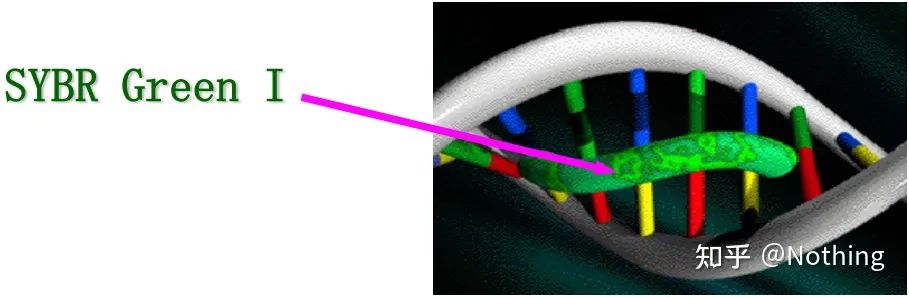
SYBR Green Ⅰ ຜູກມັດກັບຮ່ອງຂອງ DNA
ວິທີການສືບສວນTaqman probe ແມ່ນການສືບສວນ hydrolysis ທີ່ໃຊ້ທົ່ວໄປທີ່ສຸດ.ມີກຸ່ມ fluorescent ຢູ່ທີ່ 5′ ໃນຕອນທ້າຍຂອງ probe, ປົກກະຕິແລ້ວ FAM, ແລະ probe ຕົວຂອງມັນເອງເປັນລໍາດັບທີ່ສົມບູນກັບ gene ເປົ້າຫມາຍ.ມີກຸ່ມ fluorescent quenching ຢູ່ປາຍ 3′.ອີງຕາມຫຼັກການຂອງ fluorescence resonance ການໂອນພະລັງງານ (Förster resonance ໂອນພະລັງງານ, FRET), ໃນເວລາທີ່ນັກຂ່າວ fluorescent ກຸ່ມ (ໂມເລກຸນ fluorescent ຜູ້ໃຫ້ທຶນ) ແລະກຸ່ມ fluorescent quenching (ໂມເລກຸນ fluorescent ຍອມຮັບ) ມີຄວາມຕື່ນເຕັ້ນໃນເວລາທີ່ spectra overlap ແລະໄລຍະຫ່າງແມ່ນໃກ້ຊິດຫຼາຍ (7-10nm), ການຍອມຮັບຂອງ donoreculation ໂມເລກຸນ excitation ໄດ້. , ໃນຂະນະທີ່ autofluorescence ອ່ອນລົງ.ດັ່ງນັ້ນ, ໃນຕອນເລີ່ມຕົ້ນຂອງປະຕິກິລິຍາ PCR, ໃນເວລາທີ່ probe ແມ່ນບໍ່ເສຍຄ່າແລະ intact ໃນລະບົບ, ກຸ່ມ fluorescent ນັກຂ່າວຈະບໍ່ປ່ອຍ fluorescence.ໃນເວລາທີ່ annealing, primer ແລະ probe ຜູກມັດກັບແມ່ແບບ.ໃນລະຫວ່າງຂັ້ນຕອນການຂະຫຍາຍ, polymerase ສືບຕໍ່ສັງເຄາະຕ່ອງໂສ້ໃຫມ່.DNA polymerase ມີກິດຈະກໍາ 5′-3′ exonuclease.ເມື່ອເຖິງການສືບສວນ, DNA polymerase ຈະ hydrolyze probe ຈາກແມ່ແບບ, ແຍກກຸ່ມ fluorescent ນັກຂ່າວອອກຈາກກຸ່ມ fluorescent quencher, ແລະປ່ອຍສັນຍານ fluorescent.ເນື່ອງຈາກມີການພົວພັນຫນຶ່ງຕໍ່ຫນຶ່ງລະຫວ່າງ probe ແລະແມ່ແບບ, ວິທີການ probe ດີກວ່າວິທີການຍ້ອມສີໃນແງ່ຂອງຄວາມຖືກຕ້ອງແລະຄວາມອ່ອນໄຫວຂອງການທົດສອບ.ວິທີການ probe ສ່ວນໃຫຍ່ແມ່ນໃຊ້ໃນການວິນິດໄສ.
ຖາມ: ປະລິມານຢ່າງແທ້ຈິງແມ່ນຫຍັງ?Relative Quantification ແມ່ນຫຍັງ?
ຄໍາຕອບ: ປະລິມານຢ່າງແທ້ຈິງຫມາຍເຖິງການຄິດໄລ່ຈໍານວນສໍາເນົາເບື້ອງຕົ້ນຂອງຕົວຢ່າງທີ່ຈະທົດສອບໂດຍ qPCR ເຊັ່ນວ່າມີເຊື້ອໄວຣັສ HBV ຫຼາຍປານໃດໃນເລືອດ 1ml.ຜົນໄດ້ຮັບທີ່ໄດ້ຮັບຈາກປະລິມານທີ່ກ່ຽວຂ້ອງແມ່ນການປ່ຽນແປງຂອງຈໍານວນ gene ເປົ້າຫມາຍໃນຕົວຢ່າງສະເພາະໃດຫນຶ່ງທີ່ກ່ຽວຂ້ອງກັບຕົວຢ່າງອ້າງອີງອື່ນ, ແລະການສະແດງອອກຂອງ gene ແມ່ນຂຶ້ນ-regulated ຫຼືຫຼຸດລົງ-regulated.
ຖາມ: ປະລິມານການສະກັດເອົາ RNA, ປະສິດທິພາບການຖອດຂໍ້ຄວາມແບບປີ້ນກັບກັນ, ແລະປະສິດທິພາບການຂະຫຍາຍຈະສົ່ງຜົນກະທົບຕໍ່ຜົນການທົດລອງບໍ?
ຖາມ: ການເກັບຮັກສາຕົວຢ່າງ, ທາດສະກັດຈາກສານສະກັດ, ທາດປະຕິກອນການຖອດຂໍ້ຄວາມແບບປີ້ນກັບ, ແລະເຄື່ອງບໍລິໂພກທີ່ສົ່ງແສງສົ່ງຜົນກະທົບຕໍ່ຜົນການທົດລອງບໍ?
ຖາມ: ວິທີການໃດທີ່ສາມາດແກ້ໄຂຂໍ້ມູນການທົດລອງໄດ້?
ກ່ຽວກັບບັນຫາເຫຼົ່ານີ້, ພວກເຮົາຈະອະທິບາຍໃຫ້ເຂົາເຈົ້າຢ່າງລະອຽດໃນພາກກ້າວຫນ້າແລະກ້າວຫນ້າທາງດ້ານຂ້າງລຸ່ມນີ້.
2. ຄວາມຮູ້ຂັ້ນສູງ
ກ່ຽວກັບ PCR ປະລິມານ fluorescent ໃນເວລາທີ່ແທ້ຈິງ, ພວກເຮົາຕ້ອງຮັບຮູ້ຄວາມເປັນຈິງທີ່ຫລາຍພັນເອກະສານການຄົ້ນຄວ້າວິທະຍາສາດໄດ້ຖືກຈັດພີມມາໃນແຕ່ລະປີ, ໃນນັ້ນເຕັກໂນໂລຊີ PCR ປະລິມານ fluorescent ບໍ່ແມ່ນຈໍານວນຂະຫນາດນ້ອຍ.
ຖ້າບໍ່ມີມາດຕະຖານທົ່ວໄປໃນການວັດແທກການທົດລອງ PCR ປະລິມານ fluorescent, ຜົນໄດ້ຮັບອາດຈະແຕກຕ່າງກັນຢ່າງກວ້າງຂວາງ.ສໍາລັບ gene ດຽວກັນຂອງຊະນິດດຽວກັນ, ດ້ວຍວິທີການປຸງແຕ່ງດຽວກັນ, ຜົນໄດ້ຮັບການກວດພົບຍັງຈະແຕກຕ່າງກັນຢ່າງກວ້າງຂວາງ, ແລະມັນຈະເປັນການຍາກສໍາລັບຜູ້ມາຊ້າທີ່ຈະເຮັດຊ້ໍາຜົນໄດ້ຮັບດຽວກັນ.ເຈົ້າບໍ່ມີໃຜຮູ້ວ່າອັນໃດຖືກ ແລະອັນໃດຜິດ.
ນີ້ຫມາຍຄວາມວ່າ PCR ປະລິມານ fluorescent ເປັນເທກໂນໂລຍີ cheat ຫຼືເຕັກໂນໂລຢີທີ່ບໍ່ຫນ້າເຊື່ອຖືບໍ?ບໍ່ແມ່ນ, ມັນແມ່ນຍ້ອນວ່າ PCR ປະລິມານ fluorescent ມີຄວາມອ່ອນໄຫວແລະຖືກຕ້ອງຫຼາຍ, ແລະການປະຕິບັດທີ່ຜິດພາດເລັກນ້ອຍຈະໃຫ້ຜົນໄດ້ຮັບກົງກັນຂ້າມທັງຫມົດ.ການສູນເສຍເລັກນ້ອຍແມ່ນເປັນພັນໄມຫ່າງ.ຜູ້ຂຽນຂອງບົດຄວາມອາດຈະຖືກທໍລະມານເລື້ອຍໆໂດຍຜູ້ທົບທວນ.ໃນເວລາດຽວກັນ, ນັກທົບທວນຂອງວາລະສານຍັງມີຄວາມຫຍຸ້ງຍາກທີ່ຈະເລືອກເອົາຈາກຜົນການທົດລອງທີ່ແຕກຕ່າງກັນ.
ທັງໝົດ, ຊີ້ໃຫ້ເຫັນເຖິງການຂາດຄວາມເຫັນດີເຫັນພ້ອມໃນການທົດລອງ PCR ໃນເວລາຈິງ.ເພື່ອເຮັດສິ່ງນີ້, ນັກວິທະຍາສາດຊັ້ນສູງໃນອຸດສາຫະກໍາໄດ້ເລີ່ມຕົ້ນສ້າງມາດຕະຖານ,ຮຽກຮ້ອງໃຫ້ຜູ້ປະກອບສ່ວນສະໜອງຂໍ້ມູນການທົດລອງ ແລະລາຍລະອຽດການປະມວນຜົນທີ່ຈໍາເປັນບາງຢ່າງ (ລວມທັງຂໍ້ມູນທີ່ຈໍາເປັນ) ໃນບົດຄວາມເພື່ອໃຫ້ໄດ້ມາດຕະຖານເຫຼົ່ານີ້.
ຜູ້ທົບທວນສາມາດຕັດສິນຄຸນນະພາບຂອງການທົດລອງໂດຍການອ່ານລາຍລະອຽດເຫຼົ່ານີ້;ຜູ້ອ່ານໃນອະນາຄົດຍັງສາມາດໃຊ້ສິ່ງນີ້ເພື່ອເຮັດການທົດລອງອີກຄັ້ງຫຼືປັບປຸງການທົດລອງ.ຫຼັງຈາກນັ້ນ, ຜົນໄດ້ຮັບການທົດລອງທີ່ໄດ້ຮັບໃນວິທີການນີ້ແມ່ນເຕັມໄປດ້ວຍຂໍ້ມູນ, ຄຸນນະພາບສູງ, ແລະສາມາດນໍາໃຊ້ໄດ້.
MIBBI (ຂໍ້ມູນຕໍາ່ສຸດທີ່ສໍາລັບການສືບສວນດ້ານຊີວະວິທະຍາແລະຊີວະການແພດ -http://www.mibbi.org) ໄດ້ກາຍເປັນ.MIBBI ແມ່ນໂຄງການທີ່ສະຫນອງມາດຕະຖານສໍາລັບການທົດລອງ.ມັນຖືກຕີພິມໃນລັກສະນະ.ໂຄງການນີ້ແມ່ນແນໃສ່ການທົດລອງທາງຊີວະວິທະຍາຕ່າງໆ, ລວມທັງຊີວະວິທະຍາຂອງເຊນ, Microarray, qPCR ທີ່ພວກເຮົາຈະສົນທະນາໃນປັດຈຸບັນ, ແລະອື່ນໆ, ແລະສະຫນອງສໍາລັບແຕ່ລະປະເພດຂອງການທົດລອງໃນເວລາທີ່ສົ່ງຫນັງສືໃບລານ.ຂໍ້ມູນນັ້ນຄວນຈະຖືກສະໜອງໃຫ້ຕະຫຼອດເວລາ.
ໃນໂຄງການ MIBBI, ມີສອງບົດຄວາມທີ່ກ່ຽວຂ້ອງກັບ PCR ປະລິມານ fluorescent, ຄື:
· RDML (Real-Time PCR Data Markup Language) – ພາສາທີ່ມີໂຄງສ້າງ ແລະຄູ່ມືການລາຍງານສໍາລັບຂໍ້ມູນ PCR ປະລິມານທີ່ແທ້ຈິງ;
· MIQE (ຂໍ້ມູນຕໍາ່ສຸດທີ່ສໍາລັບການພິມເຜີຍແຜ່ການທົດລອງ PCR ໃນເວລາຈິງປະລິມານ) – ຂໍ້ມູນຕໍາ່ສຸດທີ່ສໍາລັບການເຜີຍແຜ່ບົດຄວາມກ່ຽວກັບການທົດລອງ PCR ປະລິມານທີ່ແທ້ຈິງ.
ທໍາອິດ, ໃຫ້ເວົ້າກ່ຽວກັບ RDML, ສະເພາະຄໍາສັບ.
ຖ້າບໍ່ມີຄໍານິຍາມມາດຕະຖານສໍາລັບທຸກສິ່ງທຸກຢ່າງ, ມັນເປັນໄປບໍ່ໄດ້ທີ່ຈະສືບຕໍ່ການສົນທະນາ, ນັ້ນແມ່ນເຫດຜົນທີ່ວ່າຄໍາອະທິບາຍຂອງຂໍ້ກໍານົດແມ່ນມີຄວາມສໍາຄັນຫຼາຍໃນການສອບເສັງ.
ຄໍາສັບທີ່ໃຊ້ໃນການທົດລອງ PCR ປະລິມານ fluorescent ປະກອບມີເນື້ອໃນດັ່ງຕໍ່ໄປນີ້.QIAGEN ໄດ້ເຮັດບົດສະຫຼຸບທີ່ດີທີ່ສຸດສໍາລັບພວກເຮົາ.ຕໍ່ໄປນີ້ແມ່ນແຫ້ງທັງຫມົດສິນຄ້າ .
ເສັ້ນໂຄ້ງການຂະຫຍາຍ
ເສັ້ນໂຄ້ງຂະຫຍາຍໃຫຍ່ໝາຍເຖິງເສັ້ນໂຄ້ງທີ່ເຮັດໃນລະຫວ່າງຂະບວນການ PCR, ໂດຍມີຕົວເລກຮອບວຽນເປັນ abscissa ແລະຄວາມເຂັ້ມຂຸ້ນຂອງ fluorescence ໃນເວລາຈິງໃນລະຫວ່າງການຕິກິຣິຍາຕາມຄຳສັ່ງ.
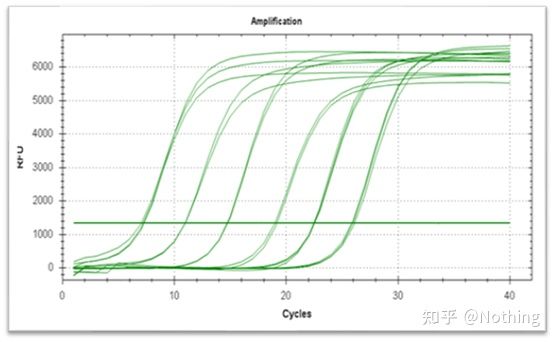
ເສັ້ນໂຄ້ງຂະຫຍາຍສຽງທີ່ດີເລີດຄວນມີລັກສະນະດັ່ງຕໍ່ໄປນີ້: ເສັ້ນພື້ນຖານແມ່ນຮາບພຽງ ຫຼື ຫຼຸດລົງເລັກນ້ອຍ, ແລະ ບໍ່ມີທ່າອ່ຽງເພີ່ມຂຶ້ນຢ່າງຈະແຈ້ງ;ຈຸດ inflection ຂອງເສັ້ນໂຄ້ງແມ່ນຈະແຈ້ງ, ແລະຄວາມຊັນຂອງໄລຍະ exponential ແມ່ນອັດຕາສ່ວນກັບປະສິດທິພາບການຂະຫຍາຍ.ຄວາມເນີນສູງຫຼາຍ, ປະສິດທິພາບການຂະຫຍາຍໃຫຍ່ຂື້ນ;ເສັ້ນໂຄ້ງຂອງ amplification ໂດຍລວມ ການຂະຫນານແມ່ນດີ, ສະແດງໃຫ້ເຫັນວ່າປະສິດທິພາບການຂະຫຍາຍຂອງແຕ່ລະທໍ່ແມ່ນຄ້າຍຄືກັນ;ໄລຍະ exponential ຂອງເສັ້ນໂຄ້ງການຂະຫຍາຍຂອງຕົວຢ່າງທີ່ມີຄວາມເຂັ້ມຂຸ້ນຕໍ່າແມ່ນເຫັນໄດ້ຊັດເຈນ.
ເສັ້ນພື້ນຖານ (Baseline)
ພື້ນຖານແມ່ນລະດັບສຽງຂອງວົງຈອນຕົ້ນ, ປົກກະຕິແລ້ວການວັດແທກລະຫວ່າງຮອບວຽນທີ 3 ແລະ 15, ເນື່ອງຈາກວ່າມູນຄ່າ fluorescence ເພີ່ມຂຶ້ນທີ່ເກີດຈາກຜະລິດຕະພັນການຂະຫຍາຍບໍ່ສາມາດກວດພົບໃນລະຫວ່າງໄລຍະເວລານີ້.ຈໍານວນຂອງຮອບວຽນທີ່ໃຊ້ໃນການຄິດໄລ່ພື້ນຖານສາມາດແຕກຕ່າງກັນແລະອາດຈະຈໍາເປັນຕ້ອງໄດ້ຫຼຸດລົງຖ້າຫາກວ່າປະລິມານແມ່ແບບສູງຖືກນໍາໃຊ້ຫຼືຖ້າຫາກວ່າລະດັບການສະແດງອອກຂອງ gene ເປົ້າຫມາຍແມ່ນສູງ.
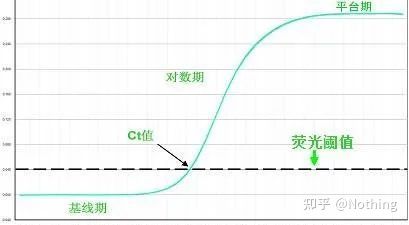
ການຕັ້ງຄ່າເສັ້ນພື້ນຖານຮຽກຮ້ອງໃຫ້ມີການເບິ່ງຂໍ້ມູນ fluorescence ຈາກເສັ້ນໂຄ້ງຂະຫຍາຍເສັ້ນ.ເສັ້ນພື້ນຖານຖືກຕັ້ງໄວ້ເພື່ອໃຫ້ການຂະຫຍາຍຕົວຂອງເສັ້ນໂຄ້ງການຂະຫຍາຍເລີ່ມຕົ້ນດ້ວຍຕົວເລກຮອບວຽນທີ່ໃຫຍ່ກວ່າຕົວເລກເທິງຂອງຮອບວຽນພື້ນຖານ.ພື້ນຖານຈໍາເປັນຕ້ອງໄດ້ຖືກກໍານົດເປັນສ່ວນບຸກຄົນສໍາລັບແຕ່ລະລໍາດັບເປົ້າຫມາຍ.ຄ່າ fluorescence ສະເລ່ຍທີ່ກວດພົບໃນຮອບທໍາອິດຈໍາເປັນຕ້ອງໄດ້ຮັບການຫັກລົບຈາກຄ່າ fluorescence ທີ່ໄດ້ຮັບໃນຜະລິດຕະພັນຂະຫຍາຍ.ເວີຊັນຫຼ້າສຸດຂອງຊອບແວ PCR ໃນເວລາຈິງຕ່າງໆ ອະນຸຍາດໃຫ້ມີການເພີ່ມປະສິດທິພາບອັດຕະໂນມັດຂອງການຕັ້ງຄ່າພື້ນຖານສໍາລັບຕົວຢ່າງສ່ວນບຸກຄົນ.
ໃນລະຫວ່າງສອງສາມຮອບທໍາອິດຂອງປະຕິກິລິຍາຂະຫຍາຍ PCR, ສັນຍານ fluorescence ບໍ່ປ່ຽນແປງຫຼາຍ.ການເຂົ້າຫາເສັ້ນຊື່ແມ່ນເອີ້ນວ່າເສັ້ນພື້ນຖານ, ແຕ່ຖ້າພວກເຮົາເບິ່ງຢ່າງໃກ້ຊິດໃນສອງສາມຮອບທໍາອິດ, ພວກເຮົາເຫັນວ່າພາຍໃນເສັ້ນພື້ນຖານແມ່ນສິ່ງທີ່ເກີດຂື້ນໃນຮູບຂ້າງລຸ່ມນີ້.
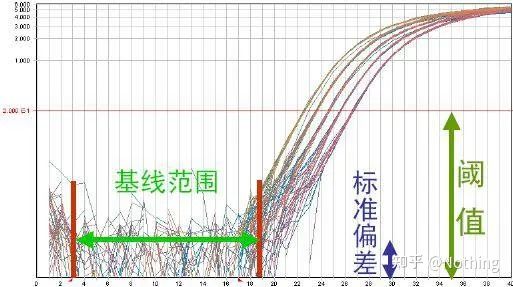
ພື້ນຖານຄວາມເປັນມາຫມາຍເຖິງ
ຄ່າ fluorescence ທີ່ບໍ່ສະເພາະໃນປະຕິກິລິຍາ .ຕົວຢ່າງ: fluorescence quenching inefficient;ຫຼືຈໍານວນຫລາຍຂອງແມ່ແບບ DNA ທີ່ມີສາຍສອງອັນເນື່ອງຈາກການນໍາໃຊ້ SYBR Green.ອົງປະກອບພື້ນຖານຂອງສັນຍານຖືກຖອດອອກທາງຄະນິດສາດໂດຍລະບົບຊອບແວ PCR ໃນເວລາຈິງ.
ສັນຍານນັກຂ່າວ
ສັນຍານນັກຂ່າວໝາຍເຖິງສັນຍານ fluorescent ທີ່ສ້າງຂຶ້ນໂດຍ SYBR Green ຫຼື fluorescently labeled probes ໃນໄລຍະ Real-Time PCR.
ສັນຍານນັກຂ່າວປົກກະຕິ (RN)
RN ຫມາຍເຖິງຄວາມເຂັ້ມ fluorescence ຂອງສີຍ້ອມຜ້ານັກຂ່າວແບ່ງອອກໂດຍຄວາມເຂັ້ມ fluorescence ຂອງສີຍ້ອມອ້າງອີງ passive ວັດແທກໃນແຕ່ລະຮອບ.
Passive Reference Dye
ໃນບາງ PCRs ເວລາຈິງ,ສີຍ້ອມ fluorescent ROX ຖືກນໍາໃຊ້ເປັນການອ້າງອິງພາຍໃນເພື່ອເຮັດໃຫ້ສັນຍານ fluorescent ເປັນປົກກະຕິ.ມັນແກ້ໄຂການປ່ຽນແປງເນື່ອງຈາກທໍ່ທີ່ບໍ່ຖືກຕ້ອງ, ຕໍາແຫນ່ງດີ, ແລະການເຫນັງຕີງຂອງ fluorescence ບົນພື້ນຖານທີ່ດີ.

ເກນ fluorescence (ເກນ)
ໄດ້ຖືກປັບຢູ່ເຫນືອມູນຄ່າພື້ນຫລັງແລະຕ່ໍາກວ່າມູນຄ່າພູພຽງຂອງເສັ້ນໂຄ້ງການຂະຫຍາຍ.ມັນຕ້ອງນອນຢູ່ໃນເຂດເສັ້ນເສັ້ນຂອງເສັ້ນໂຄ້ງຂະຫຍາຍ, ເຊິ່ງເປັນຕົວແທນຂອງລະດັບ log-linear ຂອງການກວດຫາ PCR.ຂອບເຂດຄວນຈະຖືກຕັ້ງຢູ່ໃນມຸມເບິ່ງເສັ້ນໂຄ້ງຂອງ log-amplification ເພື່ອໃຫ້ໄລຍະຂອງ log-linear ຂອງ PCR ສາມາດກໍານົດໄດ້ງ່າຍ.ຖ້າມີ genes ເປົ້າຫມາຍຫຼາຍໃນ Real-Time PCR, ຕ້ອງກໍານົດຂອບເຂດສໍາລັບແຕ່ລະເປົ້າຫມາຍ .ໂດຍທົ່ວໄປແລ້ວ, ສັນຍານ fluorescence ຂອງ 15 ຮອບທໍາອິດຂອງປະຕິກິລິຍາ PCR ຖືກນໍາໃຊ້ເປັນສັນຍານພື້ນຫລັງ fluorescence, ແລະຂອບເຂດ fluorescence ແມ່ນ 10 ເທົ່າຂອງມາດຕະຖານ deviation ຂອງສັນຍານ fluorescence ຂອງ PCR 3 ຫາ 15 ຮອບທໍາອິດ, ແລະຂອບເຂດ fluorescence ຖືກກໍານົດໃນໄລຍະການຂະຫຍາຍ PCR.ໂດຍທົ່ວໄປ, ແຕ່ລະເຄື່ອງມືມີຂອບເຂດ fluorescence ຂອງມັນທີ່ກໍານົດໄວ້ກ່ອນທີ່ຈະໃຊ້.
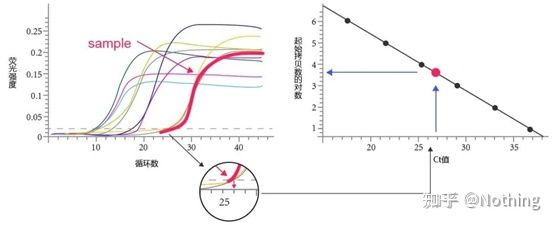
Cycle Threshold (CT) ຫຼື ຈຸດຂ້າມ (CP)
ວົງຈອນທີ່ເສັ້ນໂຄ້ງການຂະຫຍາຍຂ້າມຜ່ານຂອບເຂດ (ເຊັ່ນ: ຈຸດທີ່ກວດພົບ fluorescence ເພີ່ມຂຶ້ນຢ່າງຫຼວງຫຼາຍ).CT ສາມາດເປັນສ່ວນຫນຶ່ງແລະຈໍານວນແມ່ແບບເລີ່ມຕົ້ນສາມາດຄິດໄລ່ໄດ້.ຄ່າ CT ສະແດງເຖິງຈໍານວນຂອງຮອບວຽນທີ່ມີປະສົບການເມື່ອສັນຍານ fluorescent ໃນແຕ່ລະທໍ່ປະຕິກິລິຍາ PCR ຮອດເກນທີ່ກໍານົດໄວ້.ມີຄວາມສຳພັນເສັ້ນຊື່ລະຫວ່າງຄ່າ CT ຂອງແຕ່ລະແມ່ແບບ ແລະ logarithm ຂອງຕົວເລກສຳເນົາເບື້ອງຕົ້ນຂອງແມ່ແບບ,ຈໍານວນສໍາເນົາເບື້ອງຕົ້ນສູງຂຶ້ນ, ຄ່າ CT ຂະຫນາດນ້ອຍກວ່າ, ແລະໃນທາງກັບກັນ.ເສັ້ນໂຄ້ງມາດຕະຖານສາມາດເຮັດໄດ້ໂດຍການໃຊ້ມາດຕະຖານທີ່ມີຈໍານວນສໍາເນົາເບື້ອງຕົ້ນທີ່ຮູ້ຈັກ, ໃນນັ້ນ abscissa ເປັນຕົວແທນຂອງຄ່າ CT, ແລະ ordinate ເປັນຕົວແທນຂອງ logarithm ຂອງຈໍານວນສໍາເນົາເບື້ອງຕົ້ນ.ດັ່ງນັ້ນ, ຕາບໃດທີ່ຄ່າ CT ຂອງຕົວຢ່າງທີ່ບໍ່ຮູ້ຈັກແມ່ນໄດ້ຮັບ, ຈໍານວນສໍາເນົາເບື້ອງຕົ້ນຂອງຕົວຢ່າງສາມາດຖືກຄິດໄລ່ຈາກເສັ້ນໂຄ້ງມາດຕະຖານ.
ΔCT ຄ່າ
ຄ່າ ΔCT ອະທິບາຍຄວາມແຕກຕ່າງລະຫວ່າງ gene ເປົ້າໝາຍ ແລະຄ່າ CT gene ອ້າງອີງ endogenous ທີ່ສອດຄ້ອງກັນ, ເຊັ່ນ gene ຮັກສາເຮືອນ, ແລະຖືກນໍາໃຊ້ເພື່ອປົກກະຕິປະລິມານຂອງແມ່ແບບທີ່ໃຊ້:
⇒ΔCT = CT (gene ເປົ້າຫມາຍ) – CT (gene ອ້າງອິງ endogenous)
ΔΔCT ຄ່າ
ຄ່າ ΔΔCT ອະທິບາຍຄວາມແຕກຕ່າງລະຫວ່າງຄ່າສະເລ່ຍ ΔΔCT ຂອງຕົວຢ່າງທີ່ມີຄວາມສົນໃຈ (ຕົວຢ່າງ, ຈຸລັງທີ່ຖືກກະຕຸ້ນ) ແລະຄ່າສະເລ່ຍ ΔΔCT ຂອງຕົວຢ່າງອ້າງອີງ (ຕົວຢ່າງ, ຈຸລັງທີ່ບໍ່ໄດ້ຮັບການກະຕຸ້ນ).ຕົວຢ່າງການອ້າງອິງຍັງເອີ້ນວ່າຕົວຢ່າງການປັບທຽບແລະຕົວຢ່າງອື່ນໆທັງຫມົດແມ່ນປົກກະຕິເພື່ອການວັດແທກປະລິມານທີ່ກ່ຽວຂ້ອງ:
⇒ΔΔCT = ΔCT ສະເລ່ຍ (ຕົວຢ່າງຄວາມສົນໃຈ) – ΔCT ສະເລ່ຍ (ຕົວຢ່າງອ້າງອີງ)
genes ອ້າງອິງ endogenous (genes ອ້າງອີງ endogenous)
ລະດັບການສະແດງອອກຂອງ gene ອ້າງອິງ endogenous, ເຊັ່ນ: genes ການຮັກສາເຮືອນ (genes ຮັກສາເຮືອນ), ບໍ່ແຕກຕ່າງກັນລະຫວ່າງຕົວຢ່າງ.ການປຽບທຽບຄ່າ CT ຂອງ gene ອ້າງອິງກັບ gene ເປົ້າຫມາຍເຮັດໃຫ້ລະດັບການສະແດງອອກຂອງ gene ເປົ້າຫມາຍຖືກປັບປຸງເປັນປົກກະຕິກັບຈໍານວນ RNA ຫຼື cDNA ທີ່ປ້ອນເຂົ້າ (ເບິ່ງໃນພາກ ΔCT ຂ້າງເທິງ).
genes ອ້າງອີງພາຍໃນທີ່ຖືກຕ້ອງສໍາລັບການເຊື່ອມໂຊມຂອງ RNA ທີ່ເປັນໄປໄດ້ຫຼືມີຕົວຍັບຍັ້ງເອນໄຊໃນຕົວຢ່າງ RNA, ເຊັ່ນດຽວກັນກັບການປ່ຽນແປງຂອງເນື້ອໃນ RNA, ປະສິດທິພາບການຖອດຂໍ້ຄວາມແບບປີ້ນກັບກັນ, ການຟື້ນຕົວຂອງອາຊິດນິວຄລີອິກ, ແລະການຈັດການຕົວຢ່າງ.ເພື່ອເລືອກ gene ອ້າງອີງທີ່ດີທີ່ສຸດ, ພວກເຮົາໄດ້ແກ້ໄຂສູດການຄິດໄລ່ເພື່ອອະນຸຍາດໃຫ້ເລືອກການອ້າງອີງທີ່ດີທີ່ສຸດຂຶ້ນກັບການຕັ້ງຄ່າການທົດລອງ.
ການຄວບຄຸມພາຍໃນ
ລໍາດັບການຄວບຄຸມທີ່ຖືກຂະຫຍາຍຢູ່ໃນປະຕິກິລິຍາດຽວກັນກັບລໍາດັບເປົ້າຫມາຍແລະ probed ກັບ probe ທີ່ແຕກຕ່າງກັນ (ເຊັ່ນ, ປະຕິບັດ duplex PCR).ການຄວບຄຸມພາຍໃນມັກຈະຖືກນໍາໃຊ້ເພື່ອກົດລະບຽບການຂະຫຍາຍທີ່ລົ້ມເຫລວ, ເຊັ່ນ: ເມື່ອລໍາດັບເປົ້າຫມາຍບໍ່ໄດ້ຖືກກວດພົບ.
ຕົວຢ່າງການປັບທຽບ
ຕົວຢ່າງການອ້າງອິງ (ຕົວຢ່າງ, RNA ບໍລິສຸດຈາກເສັ້ນຈຸລັງຫຼືເນື້ອເຍື່ອ) ທີ່ໃຊ້ໃນປະລິມານທີ່ກ່ຽວຂ້ອງເພື່ອປຽບທຽບຕົວຢ່າງອື່ນໆທັງຫມົດເພື່ອກໍານົດລະດັບການສະແດງອອກຂອງເຊື້ອສາຍ.ຕົວຢ່າງການປັບທຽບສາມາດເປັນຕົວຢ່າງໃດກໍ່ຕາມ, ແຕ່ປົກກະຕິແລ້ວແມ່ນການຄວບຄຸມ (ຕົວຢ່າງ, ຕົວຢ່າງທີ່ບໍ່ໄດ້ຮັບການປິ່ນປົວຫຼືຕົວຢ່າງຈາກສູນເວລາຂອງການທົດລອງ).
ການຄວບຄຸມທາງບວກ
ໃຊ້ປະຕິກິລິຍາຄວບຄຸມກັບຈໍານວນທີ່ຮູ້ຈັກຂອງແມ່ແບບ.ການຄວບຄຸມທາງບວກມັກຈະຖືກໃຊ້ເພື່ອກວດເບິ່ງວ່າຊຸດ primer ຫຼືຊຸດ primer-probe ເຮັດວຽກຢ່າງຖືກຕ້ອງແລະປະຕິກິລິຍາຖືກຕັ້ງຄ່າຢ່າງຖືກຕ້ອງ.
ບໍ່ມີການຄວບຄຸມແມ່ແບບ (NTC)
ປະຕິກິລິຍາຄວບຄຸມທີ່ປະກອບດ້ວຍອົງປະກອບທີ່ຈໍາເປັນທັງຫມົດຂອງຕິກິຣິຍາ amplification ຍົກເວັ້ນແມ່ແບບ, ເຊິ່ງມັກຈະຖືກແທນທີ່ດ້ວຍນ້ໍາ.ການນໍາໃຊ້ NTC ສາມາດຊອກຫາການປົນເປື້ອນທີ່ເກີດຈາກການປົນເປື້ອນ reagent ຫຼື DNA ຕ່າງປະເທດ, ດັ່ງນັ້ນການຮັບປະກັນຄວາມຖືກຕ້ອງແລະຄວາມຫນ້າເຊື່ອຖືຂອງຂໍ້ມູນການກວດພົບ.ການຂະຫຍາຍການຄວບຄຸມ NTC ຊີ້ໃຫ້ເຫັນເຖິງການປົນເປື້ອນ.
ບໍ່ມີການຄວບຄຸມ RT (NRT)
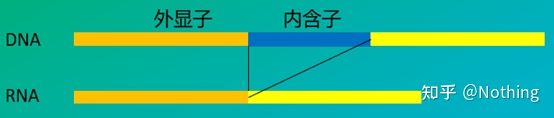
ຂະບວນການສະກັດເອົາ RNA ອາດຈະປະກອບດ້ວຍ DNA genomic ທີ່ຕົກຄ້າງ, ເຊິ່ງເປັນອັນຕະລາຍທີ່ສຸດແລະເປັນ culprit ຜົນກະທົບຕໍ່ຄຸນນະພາບຂອງຂໍ້ມູນແລະສັດຕູທໍາມະຊາດຂອງ qPCR, ສະນັ້ນໃນເວລາທີ່ການອອກແບບການທົດລອງ, ມັນຕ້ອງໄດ້ຮັບການອອກແບບພຽງແຕ່ຂະຫຍາຍການກວດພົບ RNA.ມີສອງວິທີ, ວິທີຫນຶ່ງແມ່ນການອອກແບບ primers ໃນທົ່ວ introns, ອີກຢ່າງຫນຶ່ງແມ່ນເພື່ອເອົາ DNA ອອກຫມົດ, ອັນໃດດີກວ່າ, ເຊິ່ງຈະໄດ້ປຶກສາຫາລືຕໍ່ມາ.ການຄວບຄຸມ NTR ເປັນບ່ອນແລກປ່ຽນຄວາມມະຫັດສະຈັນເພື່ອກວດຫາມົນລະພິດ DNA.ຖ້າມີການຂະຫຍາຍ, ມັນຫມາຍຄວາມວ່າມີມົນລະພິດ.
ມາດຕະຖານ
ມາດຕະຖານແມ່ນຕົວຢ່າງຂອງຄວາມເຂັ້ມຂົ້ນທີ່ຮູ້ຈັກຫຼືຕົວເລກສໍາເນົາທີ່ຖືກນໍາໃຊ້ເພື່ອສ້າງເສັ້ນໂຄ້ງມາດຕະຖານ.ເພື່ອຮັບປະກັນຄວາມຫມັ້ນຄົງຂອງມາດຕະຖານ, ຊິ້ນສ່ວນພັນທຸກໍາມັກຈະຖືກໂຄນເຂົ້າໄປໃນ plasmid ແລະຖືກນໍາໃຊ້ເປັນມາດຕະຖານ.
ເສັ້ນໂຄ້ງມາດຕະຖານ
ປົກກະຕິແລ້ວແມ່ນ diluted ເຂົ້າໄປໃນຢ່າງຫນ້ອຍ 5 gradients ຄວາມເຂັ້ມຂົ້ນກັບຜະລິດຕະພັນມາດຕະຖານຕາມອັດຕາສ່ວນສອງເທົ່າ, ແລະ 5 ຈຸດໄດ້ຖືກແຕ້ມຢູ່ໃນຈຸດປະສານງານຂອງຄ່າ CT ແລະຈໍານວນສໍາເນົາ, ແລະຈຸດແມ່ນເຊື່ອມຕໍ່ກັນເພື່ອສ້າງເສັ້ນໂຄ້ງມາດຕະຖານ.ສໍາລັບແຕ່ລະເສັ້ນໂຄ້ງມາດຕະຖານ, ຄວາມຖືກຕ້ອງຂອງມັນຕ້ອງໄດ້ຮັບການກວດສອບ.ມູນຄ່າຄວາມຊັນຢູ່ລະຫວ່າງ –3.3 ແລະ –3.8, ແລະຄວາມເຂັ້ມຂຸ້ນຂອງແຕ່ລະແມ່ນປະຕິບັດເປັນ triplicate.ຈຸດທີ່ແຕກຕ່າງຈາກຈຸດອື່ນໆຢ່າງຫຼວງຫຼາຍຄວນຖືກຍົກເລີກ.ຄ່າ CT ຂອງຕົວຢ່າງທີ່ຈະທົດສອບແມ່ນຖືກນໍາເຂົ້າໄປໃນເສັ້ນໂຄ້ງມາດຕະຖານ, ແລະລະດັບການສະແດງອອກຂອງຕົວຢ່າງທີ່ຈະທົດສອບສາມາດຄິດໄລ່ໄດ້.
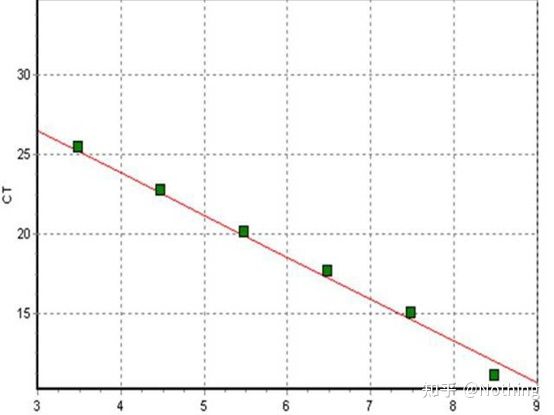
ຄ່າ CT ຂອງຕົວຢ່າງທີ່ຈະທົດສອບແມ່ນຖືກນໍາເຂົ້າໄປໃນເສັ້ນໂຄ້ງມາດຕະຖານ, ແລະຈໍານວນການຄັດລອກເບື້ອງຕົ້ນຂອງຕົວຢ່າງທີ່ຈະທົດສອບສາມາດຄິດໄລ່ໄດ້.
ປະສິດທິພາບແລະຄວາມຊັນ
ຄວາມຊັນຂອງເສັ້ນໂຄ້ງມາດຕະຖານສະແດງໃຫ້ເຫັນເຖິງປະສິດທິພາບຂອງ PCR ໃນເວລາທີ່ແທ້ຈິງ.
· A ເປີ້ນພູຂອງ -3.322 ຊີ້ໃຫ້ເຫັນວ່າປະສິດທິພາບການຂະຫຍາຍ PCR ແມ່ນ 1, ຫຼື 100% ປະສິດທິພາບ, ແລະປະລິມານຂອງຜະລິດຕະພັນ PCR ເພີ່ມຂຶ້ນສອງເທົ່າໃນແຕ່ລະວົງຈອນ.
· ຄວາມຊັນໜ້ອຍກວ່າ –3.322 (ຕົວຢ່າງ: –3.8) ສະແດງເຖິງປະສິດທິພາບ PCR
· ເປີ້ນພູທີ່ໃຫຍ່ກວ່າ –3.322 (ຕົວຢ່າງ, –3.0) ຊີ້ໃຫ້ເຫັນວ່າປະສິດທິພາບ PCR ປາກົດວ່າມີຫຼາຍກວ່າ 100%, ເຊິ່ງເປັນສິ່ງທີ່ຢາກຮູ້ຢາກເຫັນ, ວົງຈອນຂອງ PCR ສາມາດສ້າງຜະລິດຕະພັນຂະຫຍາຍໄດ້ຫຼາຍກວ່າສອງເທົ່າໄດ້ແນວໃດ?ສະຖານະການນີ້ເກີດຂື້ນໃນໄລຍະທີ່ບໍ່ແມ່ນເສັ້ນຂອງຕິກິຣິຍາ PCR, ນັ້ນແມ່ນ, ມີຈໍານວນຂະຫນາດໃຫຍ່ຂອງການຂະຫຍາຍທີ່ບໍ່ສະເພາະ.
ເສັ້ນໂຄ້ງ melting
ຫຼັງຈາກການຂະຫຍາຍ qPCR ສໍາເລັດແລ້ວ, ຜະລິດຕະພັນ PCR ໄດ້ຖືກເຮັດໃຫ້ຄວາມຮ້ອນ.ເມື່ອອຸນຫະພູມສູງຂຶ້ນ, ຜະລິດຕະພັນການຂະຫຍາຍສາຍສອງເທົ່າຄ່ອຍໆລະລາຍ, ສົ່ງຜົນໃຫ້ຄວາມເຂັ້ມຂອງ fluorescence ຫຼຸດລົງ.ໃນເວລາທີ່ອຸນຫະພູມສະເພາະໃດຫນຶ່ງ (Tm), ຈໍານວນຂອງຜະລິດຕະພັນຈະ melt.fluorescence ຫຼຸດລົງຢ່າງຫຼວງຫຼາຍ.ຜະລິດຕະພັນ PCR ທີ່ແຕກຕ່າງກັນມີມູນຄ່າ Tm ທີ່ແຕກຕ່າງກັນແລະອຸນຫະພູມການລະລາຍທີ່ແຕກຕ່າງກັນ, ດັ່ງນັ້ນຄວາມສະເພາະຂອງ PCR ສາມາດຖືກກໍານົດ.
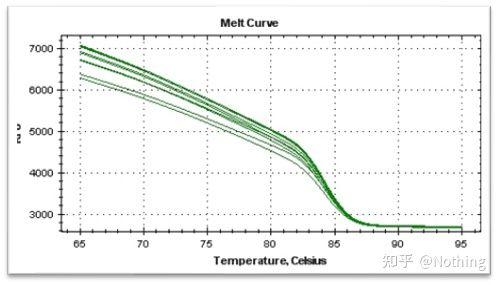
ເສັ້ນໂຄ້ງການລະລາຍ (ເສັ້ນໂຄ້ງອະນຸພັນ)
ເສັ້ນໂຄ້ງການລະລາຍແມ່ນມາຈາກການສ້າງແຜນທີ່ສູງສຸດ, ເຊິ່ງສາມາດສະແດງສະຖານະການຂອງຊິ້ນຜະລິດຕະພັນ PCR ໄດ້ຫຼາຍຂື້ນ.ນັບຕັ້ງແຕ່ອຸນຫະພູມການລະລາຍແມ່ນຄ່າ Tm ຂອງຊິ້ນ DNA, ບາງຕົວກໍານົດການທີ່ມີຜົນກະທົບຕໍ່ຄ່າ Tm ຂອງຊິ້ນ DNA ສາມາດຖືກຕັດສິນ, ເຊັ່ນ: ຂະຫນາດຊິ້ນ, ເນື້ອໃນ GC, ແລະອື່ນໆ, ເວົ້າໂດຍທົ່ວໄປແລ້ວ, ອີງຕາມຫຼັກການການອອກແບບ primer ຂອງພວກເຮົາ,ຄວາມຍາວຂອງຜະລິດຕະພັນຂະຫຍາຍຕົວຢູ່ໃນລະດັບ 80-300bp, ສະນັ້ນອຸນຫະພູມການລະລາຍຄວນຈະຢູ່ໃນລະຫວ່າງ 80 ° C ແລະ 90 ° C.
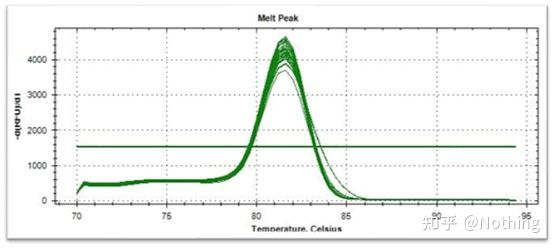
ການຕີຄວາມຫມາຍຂອງເສັ້ນໂຄ້ງ melting: ຖ້າຈຸດສູງສຸດຕົ້ນຕໍພຽງແຕ່ປາກົດລະຫວ່າງ 80 ° C-90 ° C, ມັນຫມາຍຄວາມວ່າ PCR ປະລິມານ fluorescent ແມ່ນສົມບູນ;ຖ້າຈຸດສູງສຸດຕົ້ນຕໍປາກົດຢູ່ລະຫວ່າງ 80 ° C-90 ° C ແລະຈຸດສູງສຸດອື່ນໆປະກົດວ່າຕ່ໍາກວ່າ 80 ° C, primer dimer ແມ່ນພິຈາລະນາໂດຍພື້ນຖານ.ທ່ານສາມາດພະຍາຍາມເພີ່ມອຸນຫະພູມ annealing ເພື່ອແກ້ໄຂມັນ;ຖ້າຈຸດສູງສຸດຕົ້ນຕໍປາກົດຢູ່ລະຫວ່າງ 80 ° C-90 ° C, ແລະຈຸດສູງສຸດອື່ນໆຈະປາກົດອີກເທື່ອຫນຶ່ງເມື່ອອຸນຫະພູມສູງຂຶ້ນ, ມັນໄດ້ຖືກພິຈາລະນາໂດຍພື້ນຖານແລ້ວວ່າມີການປົນເປື້ອນ DNA, ແລະ DNA ຕ້ອງໄດ້ຮັບການໂຍກຍ້າຍອອກໃນຂັ້ນຕອນທໍາອິດຂອງການທົດລອງ.
ແນ່ນອນ, ຍັງມີບາງສະຖານະການຜິດປົກກະຕິ, ເຊິ່ງຈະຖືກແບ່ງອອກຫນຶ່ງໂດຍຫນຶ່ງຂ້າງລຸ່ມນີ້.
3. ຄວາມຮູ້ຂັ້ນສູງ
ເພື່ອເຮັດ qPCR, ຂ້ອຍຕ້ອງເວົ້າວ່າ MIQE,ຂໍ້ມູນຕໍາ່ສຸດທີ່ສໍາລັບການພິມເຜີຍແຜ່ຂອງປະລິມານPCR ເວລາຈິງການທົດລອງ - ຂໍ້ມູນຕໍາ່ສຸດທີ່ສໍາລັບການເຜີຍແຜ່ບົດຄວາມກ່ຽວກັບ PCR ປະລິມານທີ່ແທ້ຈິງ - ເວລາການທົດລອງ.ເພື່ອເຮັດໃຫ້ຄວາມເຂົ້າໃຈຂອງທຸກຄົນງ່າຍຂຶ້ນ, ພວກເຮົາຈະເຮັດໃຫ້ເນື້ອໃນທີ່ສໍາຄັນງ່າຍຂຶ້ນ.
ທ່ານສາມາດຄົ້ນຫາຂໍ້ຄວາມຕົ້ນສະບັບຂອງ MIQE ໃນອິນເຕີເນັດ, ແລະສິ່ງທີ່ສໍາຄັນທີ່ສຸດແມ່ນວ່າມັນກໍານົດການບັນຊີລາຍຊື່ການກວດສອບຂໍ້ມູນທີ່ຕ້ອງໄດ້ຮັບການສະຫນອງໃຫ້ໃນເວລາທີ່ເຜີຍແຜ່ບົດຄວາມ .
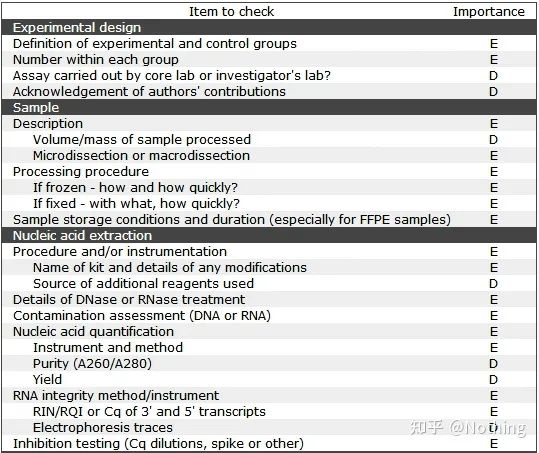
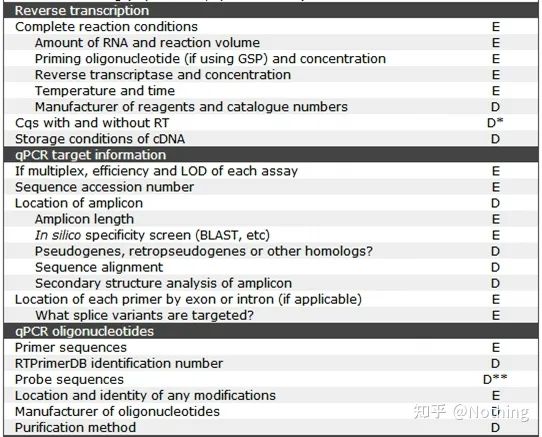
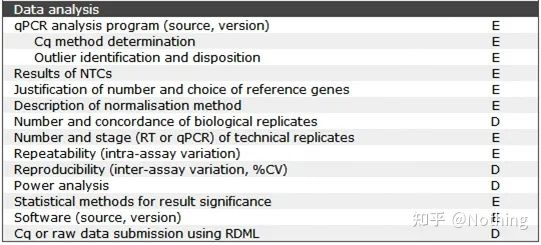
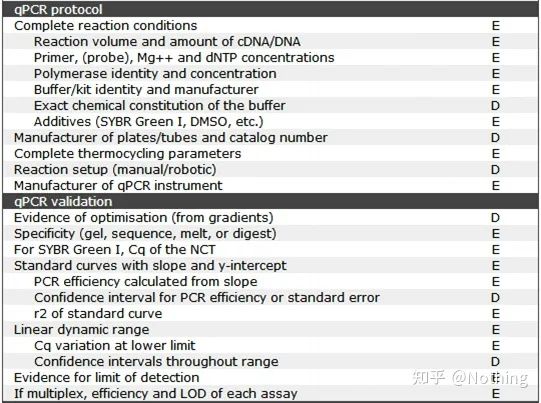
ຜູ້ທົບທວນສາມາດຕັດສິນຄຸນນະພາບຂອງການທົດລອງໂດຍການອ່ານລາຍລະອຽດເຫຼົ່ານີ້;ຜູ້ອ່ານໃນອະນາຄົດຍັງສາມາດໃຊ້ສິ່ງນີ້ເພື່ອເຮັດເລື້ມຄືນຫຼືປັບປຸງການທົດລອງ.
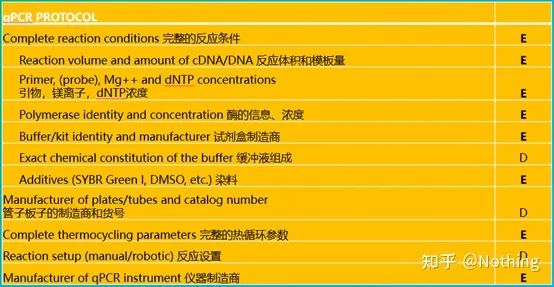
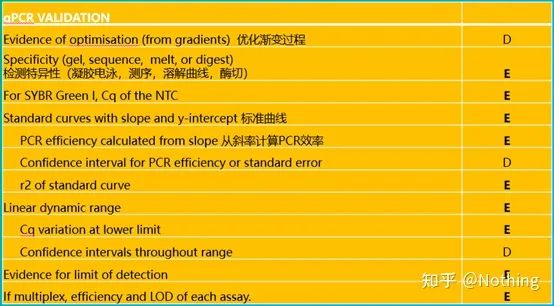
ມັນເປັນມູນຄ່າທີ່ສັງເກດວ່າໃນບັນຊີລາຍຊື່ນີ້, ຄວາມສໍາຄັນຂອງແຕ່ລະບັນຊີລາຍຊື່ແມ່ນຫມາຍດ້ວຍ E ຫຼື D ຕາມລໍາດັບ.ມັນຫມາຍຄວາມວ່າແນວໃດ?E: ຂໍ້ມູນທີ່ຈໍາເປັນ (ຕ້ອງຖືກສົ່ງ);D: ຂໍ້ມູນທີ່ຕ້ອງການ (ໃຫ້ຫຼາຍເທົ່າທີ່ເປັນໄປໄດ້).
MIQE (1)—ການອອກແບບທົດລອງ
ຄົນຂີ້ຄ້ານຫຼາຍຄົນທີ່ໄດ້ສໍາເລັດການປ້ອງກັນຂອງເຂົາເຈົ້າຫຼັງຈາກຈົບການສຶກສາຈົບການສຶກສາຂອງເຂົາເຈົ້າຈະບໍ່ຮູ້ວິທີການອອກແບບການທົດລອງເປັນເອກະລາດ, ເປີດປື້ມບັນທຶກຂອງເຂົາເຈົ້າ, ແລະເຮັດສິ່ງທີ່ອາຈານບອກໃຫ້ເຂົາເຈົ້າເຮັດ.ດ້ວຍເຫດນີ້, ການທົດລອງອອກແບບບໍ່ເຂັ້ມງວດ, ບັນນາທິການຂອງວາລະສານບອກວ່າຢາກແຕ່ງຮູບນີ້ ແລະຮູບນັ້ນ, ເຂົາເຈົ້າເຮັດແບບງຶດໆ.ນີ້ແມ່ນວິທີການເຮັດໃຫ້ຂີ້ຕົວະ!

ໃກ້ຊິດກັບບ້ານ, ຫຼັກການທໍາອິດຂອງການທົດລອງແມ່ນການກໍານົດຄວາມເຂັ້ມງວດຂອງເຫດຜົນການທົດລອງ.ສິ່ງພື້ນຖານທີ່ສຸດແມ່ນການອອກແບບທົດລອງ, ແລະສິ່ງທີ່ສໍາຄັນທີ່ສຸດຂອງການອອກແບບທົດລອງແມ່ນວິທີການກໍານົດຕົວຢ່າງເປົ້າຫມາຍ, ຕົວຢ່າງການອ້າງອິງ (ການຄວບຄຸມ), ແລະຈໍານວນການຊໍ້າຄືນ, ເພື່ອໃຫ້ຂໍ້ມູນທົດລອງສາມາດອ້າງອີງ, ປຽບທຽບ, ແລະຫນ້າເຊື່ອຖື.
ຕົວຢ່າງເປົ້າຫມາຍຫມາຍເຖິງຕົວຢ່າງທີ່ຮຽກຮ້ອງໃຫ້ພວກເຮົາກວດພົບ gene ເປົ້າຫມາຍຫຼັງຈາກການປິ່ນປົວສະເພາະໃດຫນຶ່ງ.ຕົວຢ່າງອ້າງອີງແມ່ນຕົວຢ່າງທີ່ບໍ່ມີການປິ່ນປົວໃດໆ, ເຊິ່ງມັກຈະເອີ້ນວ່າປະເພດທໍາມະຊາດໃນຊີວະສາດ.
ການທົດລອງແບບຈໍາລອງມີຄວາມສໍາຄັນຫຼາຍ.ໂດຍທົ່ວໄປແລ້ວ, ຈໍານວນຂອງການຈໍານວນທີ່ຊັກຊວນຕ້ອງມີຫຼາຍກ່ວາສາມ.ມັນເປັນສິ່ງຈໍາເປັນທີ່ຈະຈໍາແນກສິ່ງທີ່ເປັນການຈໍາລອງທາງຊີວະພາບແລະສິ່ງທີ່ເປັນການຈໍາລອງທາງດ້ານວິຊາການ.
ການຈໍາລອງທາງຊີວະພາບ: ການທົດລອງການຢັ້ງຢືນດຽວກັນເຮັດດ້ວຍວັດສະດຸທີ່ແຕກຕ່າງກັນ (ເວລາ, ພືດ, batches, ແຜ່ນຕິກິຣິຍາ).

ການຊໍ້າຊ້ອນທາງຊີວະພາບ
ໃຫ້ເຮົາໃຊ້ຢາປາບສັດຕູພືດຂອງ pepper ເປັນຕົວຢ່າງ.ພວກເຮົາຕ້ອງການສີດຢາປາບສັດຕູພືດໃສ່ສາມຕົ້ນຂອງ ABC, ຫຼັງຈາກນັ້ນສາມພືດຂອງ ABC ແມ່ນສາມ replicates ຊີວະພາບ, ແລະພວກເຂົາແມ່ນການທົດລອງການຢັ້ງຢືນດຽວກັນທີ່ດໍາເນີນດ້ວຍວັດສະດຸທີ່ແຕກຕ່າງກັນ.ແຕ່ເປັນການທົດລອງ, ການຄວບຄຸມແມ່ນຈໍາເປັນແນ່ນອນ, ດັ່ງນັ້ນພວກເຮົາສາມາດສີດຫນຶ່ງຂອງສາຂາຂອງພືດ A ເພື່ອສ້າງເປັນກຸ່ມທົດລອງຂອງພືດ A, ແລະບໍ່ສີດສາຂາອື່ນຂອງພືດ A ເພື່ອສ້າງເປັນກຸ່ມຄວບຄຸມ.ເຮັດເຊັ່ນດຽວກັນສໍາລັບ B ແລະ C.
ການຈຳລອງທາງເທັກນິກ (ການຈຳລອງທາງເທັກນິກ): ມັນເປັນການທົດລອງຊ້ໍາກັນທີ່ຖືກອອກແບບມາເພື່ອຫຼີກເວັ້ນຄວາມຜິດພາດທີ່ເກີດຈາກການດໍາເນີນງານ, ເຊິ່ງຕົວຈິງແລ້ວແມ່ນຂຸມທີ່ຊ້ໍາກັນລວມຢູ່ໃນວັດສະດຸດຽວກັນ.ການປິ່ນປົວແລະການຄວບຄຸມທັງສອງຕ້ອງມີການຕັ້ງຄ່າ replicate (ສາມຕໍາ່ສຸດທີ່) ຂອງ gene ເປົ້າຫມາຍແລະ gene ອ້າງອີງພາຍໃນ.
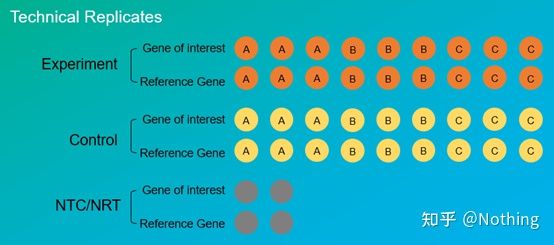
ການຄ້າງຫ້ອງທາງດ້ານວິຊາການ
ເອົາຫມາກພິກທີ່ປິ່ນປົວດ້ວຍຢາຂ້າແມງໄມ້ເປັນຕົວຢ່າງອີກເທື່ອຫນຶ່ງ.ສໍາລັບກຸ່ມທົດລອງຂອງພືດ A, ພວກເຮົາໄດ້ເຮັດສາມຮູ PCR ຂອງ 1, 2, ແລະ 3 ສໍາລັບ gene ເປົ້າຫມາຍຂອງມັນແລະ gene ອ້າງອີງພາຍໃນຕາມລໍາດັບ, ເພື່ອເອົາຄ່າສະເລ່ຍຫຼັງຈາກກວດພົບ.ສໍາລັບການຄວບຄຸມຂອງພືດ A ກຸ່ມຍັງໄດ້ຮັບການປິ່ນປົວໃນລັກສະນະດຽວກັນ.ເຊັ່ນດຽວກັນ, ເຮັດການປິ່ນປົວດຽວກັນສໍາລັບພືດ B ແລະ C.ນີ້ແມ່ນການຊໍ້າຊ້ອນທາງດ້ານວິຊາການ.
ມັນເປັນມູນຄ່າທີ່ສັງເກດວ່າສິ່ງທີ່ເຂົ້າໄປໃນສະຖິຕິແມ່ນການຄ້າງຫ້ອງທາງຊີວະພາບ, ແລະການຄ້າງຫ້ອງທາງວິຊາການແມ່ນການທົດສອບວ່າມີປະກົດການ Random ໃດໃນຂະບວນການທົດລອງ, ດັ່ງນັ້ນເພື່ອເຮັດໃຫ້ຜົນການທົດລອງມີຄວາມຫນ້າເຊື່ອຖື, ນັ້ນແມ່ນ, ເພື່ອຫຼີກເວັ້ນຄວາມຜິດພາດໂດຍການຄິດໄລ່ຄ່າສະເລ່ຍຕາມທີ່ພວກເຮົາມັກເວົ້າ.
ການຄວບຄຸມທາງລົບ—NTC ແລະ NRT
NTC (ການຄວບຄຸມບໍ່ມີແມ່ແບບ), ການຄວບຄຸມທີ່ບໍ່ມີແມ່ແບບ, ຖືກນໍາໃຊ້ເພື່ອກວດສອບວ່າອຸປະກອນການທົດລອງໄດ້ຖືກປົນເປື້ອນ.ໂດຍທົ່ວໄປ, ນ້ໍາແມ່ນໃຊ້ເປັນແມ່ແບບ.ຖ້າມີປະຕິກິລິຍາ fluorescent, ມັນຊີ້ໃຫ້ເຫັນວ່າການປົນເປື້ອນຂອງອາຊິດນິວເຄຼຍໄດ້ເກີດຂື້ນໃນຫ້ອງທົດລອງ.
ມົນລະພິດເຫຼົ່ານີ້ແມ່ນມາຈາກ: ນ້ໍາບໍ່ສະອາດ, reagents ທີ່ບໍ່ມີຄຸນສົມບັດທີ່ມີ DNA endogenous, ມົນລະພິດ primer, ມົນລະພິດອຸປະກອນຫ້ອງທົດລອງ, ມົນລະພິດ aerosol, ແລະອື່ນໆ, ຈໍາເປັນຕ້ອງໄດ້ນໍາໃຊ້ scavengers RNase ແລະ RNase inhibitors.ມົນລະພິດ Aerosol ແມ່ນມີຄວາມຫຍຸ້ງຍາກທີ່ສຸດທີ່ຈະຊອກຫາ.ຈິນຕະນາການຫ້ອງທົດລອງຂອງເຈົ້າຄືກັບຄວັນໄຟ, ມີກົດນິວຄລີອິກຕ່າງໆຖືກໂຈະຢູ່ໃນອາກາດ.

NRT (No-Reverse Transcriptase), ການຄວບຄຸມທີ່ບໍ່ມີການຖອດຂໍ້ຄວາມແບບປີ້ນກັບກັນ, ແມ່ນ RNA ທີ່ບໍ່ແມ່ນ reverse transcribed ເປັນການຄວບຄຸມທາງລົບ, ເຊິ່ງເປັນການຄວບຄຸມຂອງ gDNA residue.
ເມື່ອເຮັດການສະແດງອອກຂອງ gene, ປະລິມານຂອງ RNA ໄດ້ຖືກກວດພົບໂດຍການກວດສອບປະລິມານຂອງ cDNA ຫຼັງຈາກການຖອດຂໍ້ຄວາມຄືນ.ຖ້າມີ gDNA ຕົກຄ້າງເມື່ອ RNA ຖືກບໍລິສຸດ, ມັນຈະເຮັດໃຫ້ເກີດຄວາມຜິດພາດໃນຜົນການທົດລອງ, ເພາະວ່າຜົນໄດ້ຮັບທີ່ແທ້ຈິງທີ່ໄດ້ຮັບແມ່ນ gDNA ແລະ cDNA.ໃນລະດັບລວມ, ບໍ່ພຽງແຕ່ cDNA, gDNA ຕ້ອງໄດ້ຮັບການໂຍກຍ້າຍອອກຢ່າງສົມບູນໃນລະຫວ່າງການສະກັດເອົາ RNA.
MIQE (2) — ຂໍ້ມູນຕົວຢ່າງ
ອັນທີ່ເອີ້ນວ່າຂໍ້ມູນຕົວຢ່າງຫມາຍຄວາມວ່າເມື່ອພວກເຮົາເຜີຍແຜ່ບົດຄວາມກ່ຽວກັບ qPCR, ພວກເຮົາຕ້ອງອະທິບາຍຂໍ້ມູນຕົວຢ່າງຢ່າງຊັດເຈນ, ເຊິ່ງເປັນສ່ວນຫນຶ່ງທີ່ຂາດບໍ່ໄດ້ຂອງບົດຄວາມ.ເຊັ່ນດຽວກັນ, ເມື່ອພວກເຮົາປະມວນຜົນຕົວຢ່າງ, ພວກເຮົາຍັງຕ້ອງຄວບຄຸມການດໍາເນີນງານຂອງພວກເຮົາເອງເພື່ອຮັບປະກັນຄວາມຖືກຕ້ອງຂອງຕົວຢ່າງ.

ຄໍາອະທິບາຍຂອງຕົວຢ່າງແມ່ນພຽງແຕ່ຜົນໄດ້ຮັບ, ແລະພວກເຮົາຄວນຈະເອົາໃຈໃສ່ຫຼາຍຕໍ່ກັບວັດສະດຸທີ່ປະຕິບັດໃນລະຫວ່າງການທົດລອງທັງຫມົດ.
ການຄັດເລືອກວັດສະດຸທົດລອງ
ຕົວຢ່າງເລືອດ - ເລືອກເລືອດສົດ, ບໍ່ເກີນ 4 ຊົ່ວໂມງ.ຕົວຢ່າງເຊລ – ເລືອກເກັບເອົາເຊລສົດໃນຊ່ວງທີ່ຈະເລີນເຕີບໂຕຢ່າງແຂງແຮງ.ເນື້ອເຍື່ອສັດ—ເລືອກເນື້ອເຍື່ອທີ່ສົດ, ເຕີບໃຫຍ່ຢ່າງແຂງແຮງ.ເນື້ອເຍື່ອພືດ - ເລືອກເນື້ອເຍື່ອອ່ອນ, ສົດ.
ທ່ານຕ້ອງໄດ້ສັງເກດເຫັນວ່າມີຄໍາສໍາຄັນຢູ່ໃນສອງສາມປະໂຫຍກເຫຼົ່ານີ້: ສົດ .
ສໍາລັບຕົວຢ່າງຂ້າງເທິງ, ຊຸດທີ່ດີທີ່ສຸດ, ປະຫຍັດຄ່າໃຊ້ຈ່າຍ, ແລະຫມັ້ນຄົງໃນຕະຫຼາດແມ່ນຊຸດຂອງ Foregene, ເຊິ່ງສາມາດສະກັດ DNA ແລະ RNA ຂອງເຂົາເຈົ້າໄດ້ໄວແລະງ່າຍດາຍ.
Plant Total ຊຸດ RNA Isolation Plus
ການເກັບຮັກສາວັດສະດຸທົດລອງ
ໂດຍທົ່ວໄປແລ້ວ, ພວກເຮົາບໍ່ແນະນໍາໃຫ້ເກັບຮັກສາຕົວຢ່າງ, ຖ້າເງື່ອນໄຂອະນຸຍາດ.ຢ່າງໃດກໍຕາມ, ມີຫມູ່ເພື່ອນຫຼາຍຄົນທີ່ບໍ່ສາມາດດໍາເນີນການທົດລອງທັນທີຫຼັງຈາກການເກັບຕົວຢ່າງ, ແລະບາງຄົນກໍ່ຈໍາເປັນຕ້ອງເອົາຖັງໄນໂຕຣເຈນຂອງແຫຼວໄປພາກສະຫນາມເພື່ອເກັບຕົວຢ່າງ.
ສໍາລັບຫມູ່ທີ່ເຮັດວຽກຫນັກແບບນີ້, ຂ້ອຍເວົ້າໄດ້ວ່າເຈົ້າບໍ່ເຂົ້າໃຈການບໍລິໂພກຂອງ reagent.ໃນປັດຈຸບັນຈໍານວນຫຼາຍບໍລິສັດບໍລິໂພກ reagent ຜະລິດ reagents ທີ່ສາມາດເກັບຮັກສາຕົວຢ່າງ RNA ຢູ່ໃນອຸນຫະພູມຫ້ອງ, ແລະທ່ານສາມາດເລືອກທີ່ຈະນໍາໃຊ້ໃຫ້ເຂົາເຈົ້າ.ວິທີການເກັບຮັກສາແບບທໍາມະດາແມ່ນການເກັບຮັກສາໄນໂຕຣເຈນຂອງແຫຼວ, ໂດຍໃຊ້ຖັງນ້ໍາໄນໂຕຣເຈນຂະຫນາດນ້ອຍທີ່ງ່າຍຕໍ່ການພົກພາ.ຫຼັງຈາກທີ່ນໍາເອົາຕົວຢ່າງກັບຄືນໄປບ່ອນຫ້ອງທົດລອງ, ເກັບໄວ້ໃນຕູ້ເຢັນ -80°C.

ສໍາລັບການທົດລອງທີ່ກ່ຽວຂ້ອງກັບ RNA, ຫຼັກການຫົກຄໍາຕ້ອງປະຕິບັດຕາມ:ອຸນຫະພູມຕ່ໍາ, ບໍ່ມີ enzymes,ແລະໄວ .
ແນວຄວາມຄິດຂອງອຸນຫະພູມຕ່ໍາແມ່ນງ່າຍທີ່ຈະເຂົ້າໃຈ;ໂດຍບໍ່ມີ enzymes, RNase ແມ່ນຢູ່ທົ່ວທຸກແຫ່ງໃນໂລກທີ່ພວກເຮົາອາໄສຢູ່ (ຖ້າບໍ່ດັ່ງນັ້ນທ່ານຈະຖືກຂ້າຕາຍໂດຍ HIV), ດັ່ງນັ້ນວິທີການຫຼີກເວັ້ນ RNase ໃນເວລາທີ່ເຮັດການທົດລອງແມ່ນເປັນແນວຄວາມຄິດທີ່ສໍາຄັນຫຼາຍ;ໄວ,ບໍ່ມີ Kung Fu ໃນໂລກທີ່ບໍ່ສາມາດແຕກໄດ້, ພຽງແຕ່ຄວາມໄວບໍ່ສາມາດແຕກ.
ເພາະສະນັ້ນ, ໃນຄວາມຫມາຍ, ເວລາການສະກັດເອົາສັ້ນກວ່າ, ຊຸດທີ່ດີກວ່າ.ເປັນຫຍັງForegeneຊຸດຂອງເຄື່ອງເນັ້ນຄວາມໄວ, ເພາະວ່າພວກເຂົາຮູ້ດີ.
PS: ເດັກຍິງບາງຄົນເຮັດການທົດລອງຫຼາຍຢ່າງລະມັດລະວັງ, ແຕ່ພວກເຂົາເຈົ້າບໍ່ໄດ້ເປັນດີເປັນ slam dunk ຫຼັງຈາກການເຮັດວຽກຫຼາຍປີ.ເຂົາເຈົ້າຮູ້ສຶກວ່າພະເຈົ້າບໍ່ຍຸຕິທຳ ຈົ່ມຄົນອື່ນ ແລະຊອກຫາຊີວິດ.ໃນຄວາມເປັນຈິງ, ນາງບໍ່ເຂົ້າໃຈມັນ.ລາວບໍ່ໄດ້ປົກປ້ອງ RNA ໄດ້ດີ, ແລະຜູ້ຫຼິ້ນ slam dunk ມີຄວາມຄ່ອງແຄ້ວ.ເມື່ອລາວເຮັດການທົດລອງ, ລາວຄິດວ່າລາວຈະເຮັດການທົດລອງໃຫ້ຈົບ 3 ຄັ້ງ, ຫ້າເທື່ອ ແລະ ສອງຄັ້ງ, ແຕ່ລາວກໍເຮັດການທົດລອງໄດ້ດີ.
ຫມາຍເຫດ: ຊ້າລົງ, ໂອກາດຂອງການບຸກລຸກ RNase ຫຼາຍ.ວິທີການຝຶກອົບຮົມຕົວທ່ານເອງຈະໄວ?ບໍ່ມີທາງ, ພຽງແຕ່ປະຕິບັດຫຼາຍ.
ສໍາລັບການທົດລອງທີ່ແຕກຕ່າງກັນແລະຕົວຢ່າງທີ່ແຕກຕ່າງກັນ, ມັນຍັງມີຄວາມຈໍາເປັນທີ່ຈະອ່ານວັນນະຄະດີເພີ່ມເຕີມແລະເລືອກວິທີການທີ່ເຫມາະສົມສໍາລັບການປຸງແຕ່ງ.ສໍາລັບຂະບວນການເກັບຕົວຢ່າງແລະການເກັບຮັກສາ, MIQE ຮຽກຮ້ອງໃຫ້ມັນຕ້ອງຂຽນຢ່າງຊັດເຈນໃນກະດາດ, ເພື່ອໃຫ້ນັກທົບທວນສາມາດທົບທວນຄືນຄວາມຫນ້າເຊື່ອຖືຂອງເຈ້ຍ, ແລະມັນຍັງສະດວກສໍາລັບໄວຫນຸ່ມ stunning ເພື່ອເຮັດຊ້ໍາການທົດລອງຂອງທ່ານ.
ເຖິງແມ່ນວ່າການທົດລອງທາງຊີວະພາບແມ່ນມີຄວາມຫຍຸ້ງຍາກ, ແຕ່ພວກມັນແມ່ນລະດັບສູງ.ຖ້າທ່ານບໍ່ລະມັດລະວັງ, ທ່ານສາມາດ overturn ໂລກ.ຕົວຢ່າງ, ການເຮັດໃຫ້ SARS ເຂົ້າໄປໃນວິກິດການທາງຊີວະເຄມີ, ຫຼືການເຮັດເຂົ້າປະສົມເພື່ອຊ່ວຍປະຢັດປະຊາຊົນ 1.3 ຕື້ຄົນ.ຮູບພາບຂ້າງລຸ່ມນີ້ແມ່ນການທົດລອງທາງເຄມີ, ທ່ານຄວນເຂົ້າໃຈວ່າທ່ານພູມໃຈໃນການຄົ້ນຄວ້າຂອງທ່ານພຽງແຕ່ເບິ່ງຮູບລັກສະນະຄ້າຍຄື dick ຂອງລາວ.ລືມມັນ, ຢ່າເຮັດໃຫ້ລາວສີດໍາ.

MIQE (3) – ການສະກັດເອົາອາຊິດນິວຄລີອິກ.
ການສະກັດເອົາອາຊິດນິວເຄຼຍແມ່ນເຫດການໃຫຍ່, ແລະການທົດລອງຊີວະວິທະຍາໂມເລກຸນທັງຫມົດເລີ່ມຕົ້ນດ້ວຍການສະກັດເອົາອາຊິດນິວເຄຼຍ.ກ່ອນອື່ນ ໝົດ, ໃຫ້ຄັດລອກເນື້ອໃນຂອງ MIQE ກ່ຽວກັບການສະກັດເອົາອາຊິດນິວເຄຼຍ.
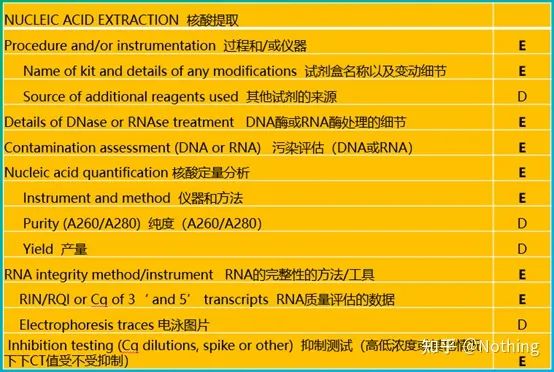
ຊອກຫາຢູ່ໃນຮູບແບບນີ້, ທ່ານບໍ່ສາມາດຢູ່ໃນຫນ້າດິນ.ແບບຟອມແມ່ນ dogma.ເພື່ອເປັນນັກຮຽນເກັ່ງ, ເຈົ້າຕ້ອງຖາມວ່າເປັນຫຍັງ.ເນື້ອໃນທີ່ສໍາຄັນຂອງຕາຕະລາງນີ້ແມ່ນ: ປະຕິບັດຕາມຄວາມບໍລິສຸດ, ຄວາມສົມບູນ, ຄວາມສອດຄ່ອງ, ແລະປະລິມານການສະກັດເອົາຂອງ RNA .
ສ່ວນທໍາອິດຂອງການຂະບວນການຫຼືເຄື່ອງມືແມ່ນຂັ້ນຕອນການສະກັດເອົາອາຊິດນິວເຄຼຍ.ຖ້າທ່ານໃຊ້ເຄື່ອງສະກັດອາຊິດນິວເຄຼຍອັດຕະໂນມັດເພື່ອສະກັດ (ກ້າວຫນ້າ, ກະລຸນາຕິດຕໍ່ຂ້ອຍເພື່ອຊື້), ທ່ານຈໍາເປັນຕ້ອງຊີ້ບອກຊື່ແບບຈໍາລອງຂອງເຄື່ອງມື.

ຊື່ຂອງຊຸດແລະ
ຊຸດໃດທີ່ຖືກນໍາໃຊ້ສໍາລັບລາຍລະອຽດການປ່ຽນແປງ, ສິ່ງທີ່ reagents ພິເສດໄດ້ຖືກເພີ່ມຫຼືສິ່ງທີ່ປະຕິບັດການພິເສດຄວນໄດ້ຮັບການອະທິບາຍຢ່າງຈະແຈ້ງເພື່ອໃຫ້ຜູ້ອື່ນສາມາດເຮັດການທົດລອງຂອງທ່ານໄດ້ຢ່າງງ່າຍດາຍ.
ບາງຄົນເພີ່ມທາດ reagents ພິເສດບາງຢ່າງໃນເວລາທີ່ສະກັດຕົວຢ່າງພິເສດ, ຄິດວ່ານີ້ແມ່ນອາວຸດລັບຂອງເຂົາເຈົ້າແລະບໍ່ໄດ້ບອກຄົນອື່ນ.ໃນຂະນະທີ່ຮັກສາຄວາມລັບ, ພວກເຂົາຍັງສູນເສຍໂອກາດທີ່ຈະເຮັດໃຫ້ບົດຄວາມຂອງເຈົ້າສະຫວ່າງ.ຢ່າສະຫລາດ, ເຈົ້າຕ້ອງມີຄວາມຊື່ສັດຫຼາຍກວ່າປະເທດເກົ່າ Zhang ໃນການຄົ້ນຄວ້າວິທະຍາສາດ, ຖ້າເຈົ້າຢາກສະຫລາດ, ບົດຄວາມຈະເຮັດໃຫ້ເຈົ້າໂງ່.
ຕ້ອງຈື່ຈໍາຈໍານວນຜະລິດຕະພັນຂອງຊຸດເມື່ອທ່ານສັ່ງຊຸດແລະຂຽນບົດຄວາມ.ໂດຍທົ່ວໄປມີສອງຕົວເລກຢູ່ໃນຊຸດ: Cat—ເລກລາຍການ (ເລກຜະລິດຕະພັນ, ເລກບົດຄວາມ), Lot — ຈໍານວນຜະລິດຕະພັນ (ໃຊ້ເພື່ອຊີ້ບອກວ່າ batch ຂອງຜະລິດຕະພັນໄດ້ມາຈາກ).
ນອກຈາກນັ້ນ, ໝາຍເລກ CAS ມັກຈະຖືກໃຊ້ໃນເວລາສັ່ງຢາຊີວະເຄມີ, ແລະຂ້ອຍຈະນິຍົມໃຊ້ກັນ.ໝາຍເລກ CAS ແມ່ນຕົວເລກທີ່ສະມາຄົມເຄມີຂອງອາເມລິກາມອບໃຫ້ແຕ່ລະຢາເຄມີໃໝ່.ໂດຍທົ່ວໄປ, ສາມຕົວເລກແມ່ນເຊື່ອມຕໍ່ໂດຍ dash.ໝາຍເລກ CAS ຂອງ Rushui: 7732-18-5.ສານເຄມີມັກຈະມີນາມແຝງຫຼາຍອັນ, ແຕ່ເລກ CAS ແມ່ນເປັນເອກະລັກ.ເມື່ອສັ່ງຢາ, ທ່ານສາມາດກວດເບິ່ງເລກ CAS ຂອງມັນກ່ອນ.
ໃກ້ຊິດກັບບ້ານ, ເປັນຫຍັງພວກເຮົາຕ້ອງອະທິບາຍສິ່ງເຫຼົ່ານີ້ຢ່າງຊັດເຈນ?ໃນຄວາມເປັນຈິງ, ມັນຍັງເປັນການກວດສອບຄຸນນະພາບຂອງການສະກັດເອົາ RNA.ການນໍາໃຊ້ເຄື່ອງມືແລະຊຸດຈະເຮັດໃຫ້ການສະກັດເອົາ RNA ສອດຄ່ອງຫຼາຍຂຶ້ນ.ຂະຫນາດການຂຸດຄົ້ນຂອງຫ້ອງທົດລອງທໍາມະດາແມ່ນບໍ່ໃຫຍ່, ແລະສາມາດໄດ້ຮັບດ້ວຍຊຸດ.
ລາຍລະອຽດຂອງການປິ່ນປົວ DNase ຫຼື RNase
ບັນຫາທີ່ສໍາຄັນຂອງ PCR ປະລິມານ fluorescent ແມ່ນການປ້ອງກັນການປົນເປື້ອນຂອງ DNA, ແລະຢ່າທົດລອງຖ້າມີການປົນເປື້ອນ.ດັ່ງນັ້ນ, ມັນເປັນສິ່ງຈໍາເປັນທີ່ຈະບອກຂະບວນການທີ່ທ່ານໃຊ້ໃນການປຸງແຕ່ງ DNA, ເພື່ອສະແດງໃຫ້ເຫັນວ່າ DNA ໃນຂະບວນການທົດລອງໄດ້ຖືກໂຍກຍ້າຍອອກຢ່າງສົມບູນແລະຫມົດແລ້ວ.ສະແດງໂດຍແຜນວາດ schematic.
ແຜນວາດແຜນວາດຂອງ RNA ແລະ DNA
ໂດຍທົ່ວໄປ, ວິທີການກໍາຈັດ DNA ແມ່ນການປິ່ນປົວ RNA ດ້ວຍ DNA ຫຼັງຈາກການສະກັດເອົາ.ຢ່າງໃດກໍຕາມ, ເຫຼົ່ານີ້ແມ່ນວິທີການທີ່ຂ້ອນຂ້າງເກົ່າ.ຊຸດສະກັດ RNA ທີ່ເປັນການຄ້າສາມາດເອົາ DNA ອອກຈາກຂະບວນການສະກັດໄດ້ໂດຍບໍ່ຕ້ອງເພີ່ມ DNase.ຕົວຢ່າງ, ຊຸດຊຸດຈາກ Foregene .
ຫມາຍເຫດ: ການຖອນ DNA ໃນລະຫວ່າງການສະກັດເອົາ RNA ແມ່ນດາບສອງດ້ານທີ່ເປັນອັນຕະລາຍຫຼາຍ, ເຊິ່ງຈະເຮັດໃຫ້ເວລາປະຕິບັດງານຂອງການສະກັດເອົາ RNA ຍາວອອກໄປແລະເພີ່ມຄວາມສ່ຽງຕໍ່ການທໍາລາຍ RNA.ໂດຍພື້ນຖານແລ້ວ, ມັນເປັນການຄ້າລະຫວ່າງຜົນຜະລິດ RNA ແລະຄວາມບໍລິສຸດ.
ນອກຈາກນັ້ນ, ປະລິມານຂອງ DNase ທີ່ເພີ່ມໃສ່ຄໍລໍາ adsorption silica ແມ່ນຫນ້ອຍຫຼາຍ, ແລະ DNase ທີ່ມີຄຸນນະພາບສູງຕ້ອງຖືກນໍາໃຊ້ເພື່ອບັນລຸຜົນກະທົບ.DNase ທີ່ບໍ່ໄດ້ຮັບການປັບປຸງບໍ່ສາມາດຍ່ອຍໄດ້ໄວ ແລະຄົບຖ້ວນ.ນີ້ແມ່ນການທົດສອບລະດັບດ້ານວິຊາການຂອງຜູ້ຄ້າ.ແນ່ນອນ, ມີພໍ່ຄ້າທີ່ແປກປະຫຼາດກວ່າທີ່ເວົ້າໂອ້ອວດວ່າ DNA ສາມາດຖືກໂຍກຍ້າຍອອກໂດຍບໍ່ມີ DNase.ມັນສາມາດເວົ້າໄດ້ວ່າຜູ້ໃດທີ່ brags ວ່າ DNA ສາມາດເອົາອອກໄດ້ຫມົດໂດຍບໍ່ມີ DNase ແມ່ນ hooligan.DNA ແມ່ນໂຄງສ້າງສອງສາຍທີ່ຂ້ອນຂ້າງຄົງທີ່, ແລະມັນບໍ່ສາມາດຖືກລົບລ້າງພຽງແຕ່ໂດຍການເວົ້າແລະຫົວ.
ການປະເມີນການປົນເປື້ອນ
ວິທີການປະເມີນ: ການກວດພົບ electrophoresis, 1% agarose, 6V / cm, 15min, loading 1-3 ul
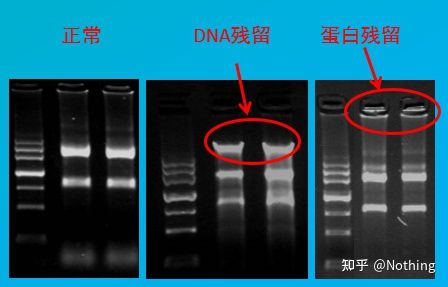
ການວິເຄາະປະລິມານອາຊິດນິວເຄຼຍ
ປົກກະຕິແລ້ວແມ່ນວັດແທກໂດຍໃຊ້ spectrophotometer UV.ທໍາອິດໃຫ້ຂ້ອຍນິຍົມຄວາມຫມາຍຂອງສາມຄ່າຂອງ OD260, OD280, ແລະ OD230.
· OD260nm: ມັນແມ່ນຄວາມຍາວຂອງການດູດຊຶມຂອງລະດັບສູງສຸດຂອງການດູດຊຶມຂອງອາຊິດນິວຄລີອິກ, ແລະຄ່າທີ່ດີທີ່ສຸດທີ່ວັດແທກໄດ້ຕັ້ງແຕ່ 0.1 ຫາ 1.0.ຖ້າບໍ່, ເຈືອຈາງຫຼືສຸມໃສ່ຕົວຢ່າງເພື່ອໃຫ້ມັນຢູ່ໃນຂອບເຂດ.
· OD280nm: ມັນແມ່ນຄວາມຍາວຂອງການດູດຊຶມຂອງລະດັບສູງສຸດຂອງການດູດຊຶມຂອງທາດໂປຼຕີນແລະສານ phenolic.
· OD230nm: ມັນແມ່ນຄວາມຍາວຂອງການດູດຊຶມຂອງຄາໂບໄຮເດດສູງສຸດຂອງການດູດຊຶມສູງສຸດ.
ຕໍ່ໄປ, ໃຫ້ເວົ້າກ່ຽວກັບບົດບາດຂອງຕົວຊີ້ວັດແຕ່ລະຄົນ.ສໍາລັບ A260, ມັນສາມາດຖືກນໍາໃຊ້ເພື່ອວັດແທກຜົນຜະລິດຂອງອາຊິດ nucleic.ເມື່ອ OD260=1, dsDNA=50μg/ml, ssDNA=37μg/ml, RNA=40μg/ml.
ເພື່ອຄວາມບໍລິສຸດ, ພວກເຮົາຈໍາເປັນຕ້ອງໄດ້ເບິ່ງອັດຕາສ່ວນທີ່ພວກເຮົາເຫັນທົ່ວໄປ: OD260/280 ແລະ OD260/230.
· DNA ບໍລິສຸດ: OD260/280 ແມ່ນປະມານເທົ່າກັບ 1.8.ເມື່ອມັນໃຫຍ່ກວ່າ 1.9, ມັນຊີ້ໃຫ້ເຫັນວ່າມີມົນລະພິດ RNA, ແລະເມື່ອມັນຫນ້ອຍກວ່າ 1.6, ມັນຊີ້ໃຫ້ເຫັນວ່າມີທາດໂປຼຕີນແລະ phenol ມົນລະພິດ.
· RNA ບໍລິສຸດ: 1.7
· OD260/230: ບໍ່ວ່າຈະເປັນ DNA ຫຼື RNA, ຄ່າອ້າງອີງແມ່ນ 2.5.ເມື່ອມັນຫນ້ອຍກວ່າ 2.0, ມັນຊີ້ໃຫ້ເຫັນວ່າມີມົນລະພິດຂອງ້ໍາຕານ, ເກືອແລະອິນຊີ.
ຄວາມສົມບູນຂອງ RNA
ມັນເປັນສິ່ງສໍາຄັນທີ່ສຸດທີ່ຈະວັດແທກຄວາມສົມບູນຂອງ RNA.ໂດຍທົ່ວໄປແລ້ວ, ມັນຈໍາເປັນຕ້ອງເຮັດການທົດລອງ RNA denaturation gel ເພື່ອກວດເບິ່ງວ່າຄວາມສະຫວ່າງລະຫວ່າງ 28S ແລະ 18S RNA ແມ່ນຄວາມສໍາພັນສອງເທົ່າ.ເມື່ອ 5S ແຖບທີສາມປາກົດ, ມັນຫມາຍຄວາມວ່າ RNA ໄດ້ເລີ່ມຕົ້ນທີ່ຈະຊຸດໂຊມ, ຍົກເວັ້ນສໍາລັບສັດທີ່ບໍ່ມີກະດູກສັນຫຼັງ.

ຂໍ້ມູນສໍາລັບການປະເມີນຄຸນນະພາບ RNA: ນອກເຫນືອຈາກການທົດສອບຂ້າງເທິງ, ຍັງມີການທົດສອບເຄື່ອງມືທີ່ກ້າວຫນ້າທາງດ້ານຄວາມສົມບູນຂອງ RNA ເຊັ່ນ: ການທົດສອບຄວາມສົມບູນ RQI ຂອງລະບົບ electrophoresis ອັດຕະໂນມັດ Experion, ເຊິ່ງສາມາດກວດພົບວ່າ RNA ຖືກທໍາລາຍໂດຍເບິ່ງເຫັນໄດ້ບໍ.
ໃນການຄົ້ນຄວ້າວິທະຍາສາດ, PCR ປະລິມານ fluorescent ແມ່ນການປຽບທຽບລະຫວ່າງ gene ເປົ້າຫມາຍແລະ gene ອ້າງອີງພາຍໃນ.ດັ່ງນັ້ນ, ໃນຂະບວນການຮັກສາຕົວຢ່າງ RNA, ການສະກັດເອົາ RNA, ແລະອື່ນໆ, ເປົ້າຫມາຍຕົ້ນຕໍແມ່ນເພື່ອຮັບປະກັນຄວາມສົມບູນຂອງ RNA.
ຄວາມສົມບູນຂອງ RNA ມີຜົນກະທົບແນວໃດຕໍ່ຄວາມສົມດູນລະຫວ່າງ gene ເປົ້າຫມາຍແລະ gene ອ້າງອີງພາຍໃນສາມາດເຂົ້າໃຈໄດ້ງ່າຍຈາກຮູບຂ້າງລຸ່ມນີ້.ການເຊື່ອມໂຊມຈະນໍາໄປສູ່ຄວາມບໍ່ສົມບູນຂອງ gene, ບໍ່ວ່າຈະເປັນຄວາມບໍ່ສົມບູນຂອງ gene ອ້າງອີງພາຍໃນຫຼືຄວາມບໍ່ສົມບູນຂອງ gene ເປົ້າຫມາຍ, ມັນຈະມີຜົນກະທົບຢ່າງຫຼວງຫຼາຍຕໍ່ຂໍ້ມູນ.

ແຜນວາດແຜນວາດຂອງ gene ເປົ້າໝາຍ ແລະ gene ອ້າງອີງ, ຈະຕ້ອງບໍ່ແມ່ນຄວາມຈິງ
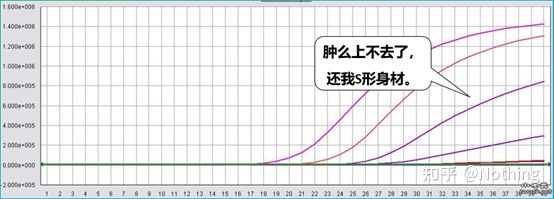
ການທົດສອບການຍັບຍັ້ງ (ບໍ່ວ່າຈະເປັນຄ່າ CT ຈະຖືກສະກັດກັ້ນພາຍໃຕ້ຄວາມເຂັ້ມຂົ້ນສູງຫຼືຕ່ໍາຫຼືເງື່ອນໄຂອື່ນໆ)
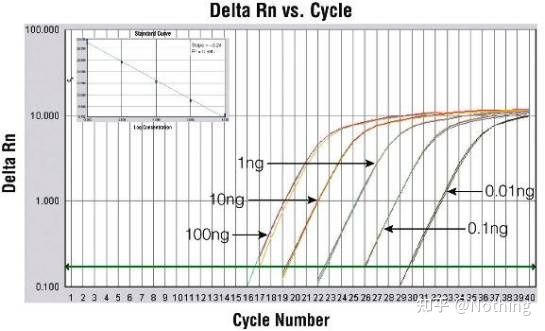
ເອົາຕົວເລກນີ້ເປັນຕົວຢ່າງ, ຄ່າ Ct ຂອງເສັ້ນໂຄ້ງຫ້າແມ່ນດັ່ງຕໍ່ໄປນີ້.ການແຜ່ກະຈາຍຂອງຄ່າ CT ລະຫວ່າງເສັ້ນໂຄ້ງແມ່ນບໍ່ສະເຫມີພາບ, ແລະຄ່າ Ct ຖືກຊັກຊ້າພາຍໃຕ້ຄວາມເຂັ້ມຂົ້ນສູງແລະຕ່ໍາ, ເຊິ່ງເປັນກໍລະນີຂອງການຍັບຍັ້ງ PCR.
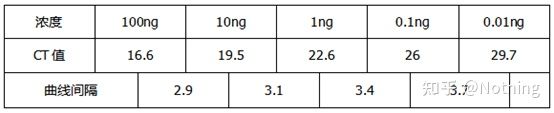
ຈຸດສໍາຄັນ: ໃນຂະບວນການສະກັດເອົາ RNA, ພວກເຮົາຈໍາເປັນຕ້ອງປະຖິ້ມຄວາມເຂົ້າໃຈຜິດແລະສ້າງຕັ້ງທີ່ຖືກຕ້ອງ.
ຄວາມຄິດທີ່ຜິດພາດແມ່ນ: ການສະກັດເອົາ RNA ພຽງແຕ່ຕິດຕາມຜົນຜະລິດ, ຄິດວ່າປະລິມານ RNA ທີ່ໄດ້ຮັບຫຼາຍ, ດີກວ່າ.ໃນຄວາມເປັນຈິງ, ເມື່ອພວກເຮົາເຮັດການວັດແທກ, ຖ້າຈໍານວນຂອງ genes ບໍ່ຫຼາຍ, ພວກເຮົາບໍ່ຕ້ອງການ RNA ຫຼາຍ.ປະລິມານຂອງ RNA ທີ່ທ່ານສະກັດອອກມາແມ່ນຫຼາຍກ່ວາພຽງພໍ .
ແນວຄວາມຄິດທີ່ຖືກຕ້ອງແມ່ນ:ການສະກັດເອົາ RNA ຄວນປະຕິບັດຕາມຄວາມບໍລິສຸດ, ຄວາມສົມບູນແລະຄວາມສອດຄ່ອງ.ຄວາມບໍລິສຸດສາມາດຮັບປະກັນວ່າການຖອດຂໍ້ຄວາມແບບປີ້ນກັບກັນຕໍ່ມາບໍ່ໄດ້ຖືກຍັບຍັ້ງແລະຂໍ້ມູນຈະບໍ່ໄດ້ຮັບຜົນກະທົບຈາກ DNA.ຄວາມຊື່ສັດຮັບປະກັນຄວາມສົມດຸນຂອງລໍາດັບເປົ້າຫມາຍແລະການອ້າງອີງພາຍໃນ.ຄວາມສອດຄ່ອງຮັບປະກັນການໂຫຼດຕົວຢ່າງທີ່ຫມັ້ນຄົງ.
MIQE (4) – ການຖອດຂໍ້ຄວາມແບບປີ້ນກັບກັນ
ຄວາມເຂົ້າໃຈຜິດ: ການສະແຫວງຫາປະລິມານຕົວຢ່າງທີ່ສູງຂຶ້ນ.
ແນວຄວາມຄິດທີ່ຖືກຕ້ອງ: ປະຕິບັດຕາມຄວາມສອດຄ່ອງ (ຄວາມຫມັ້ນຄົງ), ໂດຍບໍ່ຄໍານຶງເຖິງຈໍານວນ RNA ທີ່ຖືກໂຫລດ, ປະສິດທິພາບຂອງ reverse transcription ຍັງຄົງສອດຄ່ອງ, ຮັບປະກັນວ່າຄວາມແຕກຕ່າງຂອງ cDNA ສາມາດສະທ້ອນເຖິງຄວາມແຕກຕ່າງຂອງ mRNA ຢ່າງແທ້ຈິງ.
ພວກເຮົາອະທິບາຍຂະບວນການນີ້ດ້ວຍແຜນວາດ schematic:
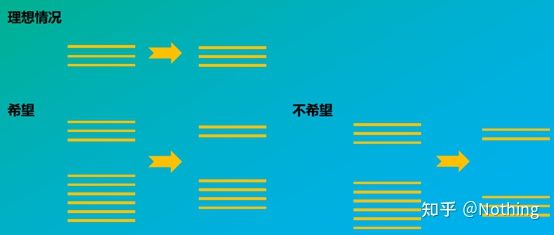
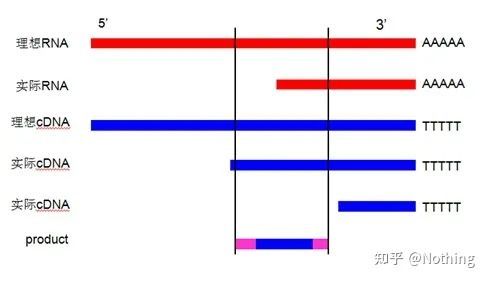
ແຜນວາດ Schematic ຂອງປະສິດທິພາບການຖອດຂໍ້ຄວາມແບບປີ້ນກັບກັນ, ບໍ່ເປັນຄວາມຈິງ
ກ່ອນອື່ນຫມົດ, ພວກເຮົາຕ້ອງໄດ້ເຂົ້າໃຈຄວາມແຕກຕ່າງລະຫວ່າງຂະບວນການ transcription reverse ແລະຂະບວນການ PCR.PCR ດໍາເນີນຂະບວນການເຮັດຄວາມຮ້ອນແລະການຫມຸນຫຼາຍ, ແລະຊິ້ນສ່ວນເປົ້າຫມາຍຈະເລີນເຕີບໂຕເປັນຕົວເລກ;ໃນຂະນະທີ່ reverse transcription ບໍ່ມີຂະບວນການນີ້, ພວກເຮົາສາມາດຈິນຕະນາການວ່າ reverse transcription ແມ່ນຕົວຈິງແລ້ວຫນຶ່ງຕໍ່ຫນຶ່ງໃນລະຫວ່າງການຂະບວນການ replication ເປັນຈໍານວນຫຼາຍຂອງ RNA.
ຍ້ອນວ່າມັນສາມາດໄດ້ຮັບຂໍ້ມູນ cDNA ຫຼາຍເທົ່າ, ມັນຄວນຈະເຂົ້າໃຈໃນປັດຈຸບັນ, ເພາະວ່າຊິ້ນສ່ວນໃຫຍ່ແລະຂະຫນາດນ້ອຍໄດ້ຖືກຖອດຖອນຄືນ, ແລະມັນເປັນໄປບໍ່ໄດ້ທີ່ຈະສຸມໃສ່ຫນຶ່ງຊິ້ນ.ແລະເນື່ອງຈາກວ່າປະລິມານຂອງ RNA ແມ່ນຂ້ອນຂ້າງຫນ້ອຍ, ຈໍານວນຂອງ cDNA ທີ່ໄດ້ຮັບແມ່ນຂ້ອນຂ້າງຫນ້ອຍ, ບໍ່ເຫມືອນກັບ PCR, ມີຜົນກະທົບການຂະຫຍາຍ, ສະນັ້ນມັນເປັນໄປບໍ່ໄດ້ໂດຍພື້ນຖານແລ້ວທີ່ຈະກວດພົບ.
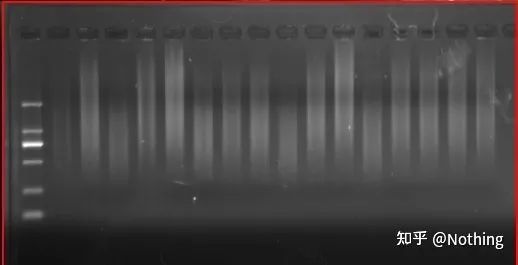
ຜົນໄດ້ຮັບຂອງ cDNA electrophoresis
ອັນທີສອງ, ໂດຍວິທີທາງການ, ການຖອດຂໍ້ຄວາມແບບປີ້ນກັບກັນແມ່ນປະຕິບັດຫນຶ່ງຕໍ່ຫນຶ່ງ, ແຕ່ບໍ່ມີ transcriptase reverse ຈາກບໍລິສັດໃດກໍ່ຕາມສາມາດບັນລຸຜົນກະທົບນີ້.ໂດຍພື້ນຖານແລ້ວ, ປະສິດທິພາບຂອງ transcriptases ປີ້ນກັບກັນຫຼາຍທີ່ສຸດ wanders ລະຫວ່າງ 30-50%.ຖ້າເປັນແບບນີ້, ພວກເຮົາດີກວ່າຈະມີປະສິດທິພາບການຖອດຂໍ້ຄວາມແບບປີ້ນກັບທີ່ຂ້ອນຂ້າງຄົງທີ່, ເຊິ່ງເປັນສິ່ງທີ່ພວກເຮົາຕ້ອງການເບິ່ງໃນຮູບ: 3 RNAs ໄດ້ຮັບ 2 cDNA, 6 RNAs ໄດ້ຮັບ 4 cDNA, ດັ່ງນັ້ນບໍ່ວ່າຕົວຢ່າງຈະໂຫຼດຫຼາຍປານໃດ, ປະສິດທິພາບການຖອດຂໍ້ຄວາມແບບປີ້ນກັບກັນແມ່ນຂ້ອນຂ້າງຄົງທີ່.ພວກເຮົາບໍ່ຕ້ອງການເບິ່ງສະຖານະການທີ່ປະສິດທິພາບການຖອດຂໍ້ຄວາມແບບປີ້ນກັບກັນແມ່ນບໍ່ສະຖຽນລະພາບແລະຄວາມເຂັ້ມຂຸ້ນສູງຖືກຍັບຍັ້ງ.
ດັ່ງນັ້ນ, ວິທີການກວດສອບວ່າປະສິດທິພາບການຖອດຂໍ້ຄວາມແບບປີ້ນກັບກັນມີຄວາມຫມັ້ນຄົງບໍ?ວິທີການແມ່ນງ່າຍດາຍຫຼາຍ, ທ່ານພຽງແຕ່ຕ້ອງການເຮັດການທົດສອບການປຽບທຽບ: ຫນຶ່ງແມ່ນເພື່ອ reverse transcript ເຂົ້າໄປໃນ cDNA ຫຼັງຈາກການເຈືອຈາງສອງເທົ່າຂອງ RNA, ແລະອີກຢ່າງຫນຶ່ງແມ່ນເພື່ອເຮັດໃຫ້ dilution ສອງເທົ່າຫຼັງຈາກ reverse transcribing ເຂົ້າໄປໃນ cDNA, ແລະຫຼັງຈາກນັ້ນເຮັດ qPCR ເພື່ອເບິ່ງຄວາມຊັນທີ່ໄດ້ຮັບມັນສອດຄ່ອງ.ໃນຖານະເປັນນັກຮຽນຊັ້ນນໍາ, ທ່ານຄວນເຂົ້າໃຈມັນພາຍໃນວິນາທີ.ດັ່ງທີ່ສະແດງຢູ່ລຸ່ມນີ້:
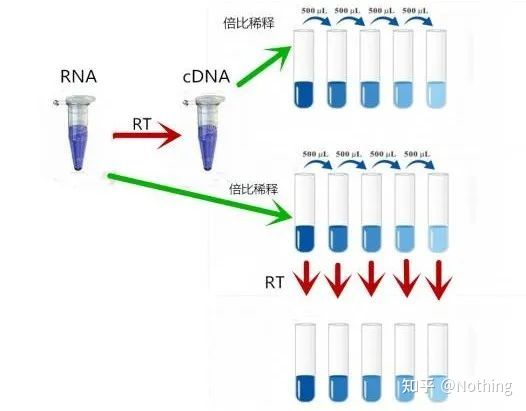
ການເຈືອຈາງຂອງ RNA ແລະ cDNA ເພື່ອທົດສອບວ່າປະສິດທິພາບຂອງການຖອດຂໍ້ຄວາມແບບປີ້ນກັບແມ່ນມີຄວາມໝັ້ນຄົງຫຼືບໍ່
Reverse transcriptase ແລະຊຸດ
ເຮັດແນວໃດ PCR ປະລິມານ fluorescent ທີ່ສົມບູນແບບມີ transcriptase ປີ້ນກັບກັນທີ່ດີເລີດແລະຊຸດ.Reverse transcriptase ແບ່ງອອກເປັນສອງປະເພດຕາມແຫຼ່ງທີ່ມາ, AMV ຫຼືM-MLV, ແລະການປະຕິບັດຂອງພວກເຂົາແມ່ນຄືກັນກັບທີ່ສະແດງຢູ່ໃນຕາຕະລາງ.
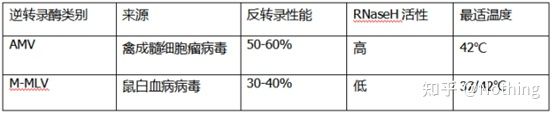
ກິດຈະກໍາ RNase H
RNase H ແມ່ນ Ribonuclease H, ຊື່ຈີນແມ່ນ ribonuclease H, ເຊິ່ງເປັນ endoribonuclease ທີ່ສາມາດ hydrolyze RNA ໂດຍສະເພາະໃນລະບົບຕ່ອງໂສ້ປະສົມ DNA-RNA.RNase H ບໍ່ສາມາດ hydrolyze ພັນທະບັດ phosphodiester ໃນ DNA ຫຼື RNA ສາຍດຽວຫຼືສອງສາຍ, ນັ້ນແມ່ນ, ມັນບໍ່ສາມາດຍ່ອຍ DNA ຫຼື RNA ສາຍດຽວຫຼືສອງສາຍ.ໃຊ້ທົ່ວໄປໃນການສັງເຄາະສາຍທີສອງຂອງ cDNA.
ມັນເປັນເລື່ອງແປກ.ພວກເຮົາເວົ້າວ່າ reverse transcriptase ມີກິດຈະກໍາ RNase H, ບໍ່ແມ່ນວ່າ reverse transcriptase ມີ RNase H, ແລະມັນອາດຈະບໍ່ສາມາດແຍກ RNase H ຈາກ reverse transcriptase ໄດ້, ບາງທີອາດເປັນຍ້ອນການສອດຄ່ອງຂອງບາງກຸ່ມໃນ reverse transcriptase ກິດຈະກໍານີ້ແມ່ນເກີດມາຈາກ reverse transcriptase.
ດັ່ງນັ້ນ, ໂດຍບໍ່ຄໍານຶງເຖິງປະສິດທິພາບການຖອດຂໍ້ຄວາມແບບປີ້ນກັບກັນທີ່ສູງຂຶ້ນຂອງ AMV, ກິດຈະກໍາ RNase H ຂອງມັນຫຼຸດລົງຜົນຜະລິດຂອງ cDNA.ແນ່ນອນ, ຜູ້ຜະລິດ reagent ກໍາລັງປັບປຸງຜະລິດຕະພັນຂອງພວກເຂົາຢ່າງຕໍ່ເນື່ອງເພື່ອລົບລ້າງກິດຈະກໍາ RNase H ໃນ reverse transcriptase ຫຼາຍເທົ່າທີ່ເປັນໄປໄດ້ເພື່ອເພີ່ມຜົນຜະລິດຂອງ cDNA.
ອຸນຫະພູມ Annealing
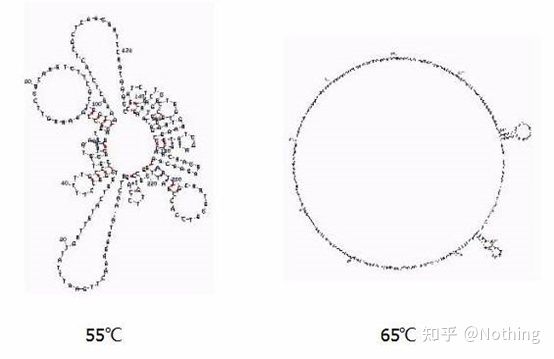
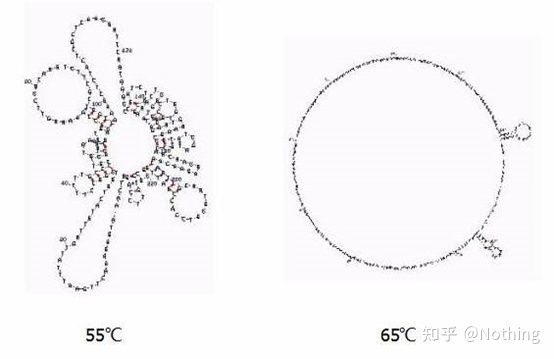
ໂຄງສ້າງຂັ້ນສອງຂອງ RNA ໃນອຸນຫະພູມທີ່ແຕກຕ່າງກັນ
ເບິ່ງຮູບຂ້າງເທິງສໍາລັບໂຄງສ້າງຮອງຂອງ RNA ໃນອຸນຫະພູມທີ່ແຕກຕ່າງກັນ, ແລະນໍາໃຊ້ເຄື່ອງມືອອນໄລນ໌ mFold ເພື່ອກໍານົດໂຄງສ້າງທີສອງຂອງຊິ້ນເປົ້າຫມາຍພາຍໃຕ້ເງື່ອນໄຂອຸນຫະພູມແລະຄວາມເຂັ້ມຂົ້ນຂອງເກືອ.ຢູ່ທີ່ 55 ° C, ໂຄງສ້າງຮອງຂອງ RNA ແມ່ນຍັງສັບສົນຫຼາຍ, reverse transcriptase ບໍ່ສາມາດເຮັດວຽກໄດ້, ແລະໂຄງສ້າງທີສອງບໍ່ສາມາດແກ້ໄຂໄດ້ຢ່າງສົມບູນຈົນກ່ວາ 65 ° C, ໃນຂະນະທີ່ອຸນຫະພູມທີ່ດີທີ່ສຸດຂອງ AMV ແລະ M-MLV ແມ່ນຕ່ໍາກວ່າອຸນຫະພູມນີ້.
ຈະເຮັດແນວໃດ?ໂຄງສ້າງຂັ້ນສອງແມ່ນການຈັບຄູ່ແບບປະສົມປະສານຂອງແມ່ແບບຕົວມັນເອງ, ເຊິ່ງນໍາໄປສູ່ການແຂ່ງຂັນທີ່ເຂັ້ມແຂງລະຫວ່າງ primer ແລະ reverse transcriptase ແລະແມ່ແບບ, ເຊິ່ງກໍ່ໃຫ້ເກີດບັນຫາຕ່າງໆເຊັ່ນ E ຕ່ໍາແລະການເຮັດເລື້ມຄືນທີ່ບໍ່ດີ.
ຈະເຮັດແນວໃດ?ພຽງແຕ່ເພີ່ມອຸນຫະພູມ annealing ຫຼາຍເທົ່າທີ່ເປັນໄປໄດ້.
ຜູ້ຜະລິດ reagent ຈໍານວນຫຼາຍກໍາລັງປັບປຸງ transcriptase ກັບຄືນຂອງເຂົາເຈົ້າໂດຍຜ່ານວິສະວະກໍາພັນທຸກໍາ.ບາງຄົນເພີ່ມອຸນຫະພູມຕິກິຣິຍາ, ເຊັ່ນ Jifan ແລະ Aidelai, ແລະບາງຄົນເອົາກຸ່ມທີ່ມີການເຄື່ອນໄຫວຂອງ enzyme RNase H ເພື່ອປັບປຸງຄວາມໃກ້ຊິດລະຫວ່າງ enzyme ແລະແມ່ແບບ RNA.ຄວາມໃກ້ຊິດສູງສາມາດແຂ່ງຂັນບີບອອກໂຄງສ້າງຮອງແລະອ່ານຜ່ານລຽບ, ແລະຍັງປັບປຸງປະສິດທິພາບຂອງການຖອດຂໍ້ຄວາມແບບປີ້ນກັບກັນຢ່າງຫຼວງຫຼາຍ.
ຈຸດສໍາຄັນ: ການຖອດຂໍ້ຄວາມແບບປີ້ນຄືນແມ່ນມີຄວາມສໍາຄັນກວ່າທີ່ຈະປະຕິບັດຕາມຄວາມສອດຄ່ອງຂອງປະສິດທິພາບການຖອດຂໍ້ຄວາມແບບປີ້ນກັບກັນ (enzymes ບໍ່ພຽງແຕ່ຈະມີປະສິດທິພາບແຕ່ຍັງມີຄວາມຫມັ້ນຄົງ), ແທນທີ່ຈະເປັນຈໍານວນຕົວຢ່າງທີ່ໂຫລດ, ຖ້າມັນບໍ່ແມ່ນ PCR ປະລິມານ fluorescent ຂະຫນາດໃຫຍ່ໂດຍສະເພາະ, ມັນຈະເປັນໄປບໍ່ໄດ້ທັງຫມົດ.ຫຼາຍ cDNAs.
ຜູ້ຜະລິດຕ່າງໆຍັງໄດ້ພະຍາຍາມບາງຢ່າງໃນການຄົ້ນຫາຄວາມສອດຄ່ອງ.ຕົວຢ່າງ, ບໍລິສັດສ່ວນໃຫຍ່ໃນປັດຈຸບັນໄດ້ຫຸ້ມຫໍ່ reverse transcription ເປັນຊຸດມາດຕະຖານສໍາລັບການຂາຍ, ເຊິ່ງເປັນທາງເລືອກທີ່ດີ.
ຕົວຢ່າງ, ຊຸດຊຸດ RT Easy Series ຂອງ Foregene:
RT Easy I (Master premix ສໍາລັບຊຸດສັງເຄາະ cDNA strand ທໍາອິດ)
MIQE (5) – ຂໍ້ມູນ gene ເປົ້າຫມາຍ

ຕົວເລກຂ້າງເທິງອະທິບາຍ
1. ບໍ່ວ່າຈະເປັນ gene ນີ້ມີປະສິດທິພາບສໍາລັບການທົດລອງຊ້ໍາກັນໂດຍທົ່ວໄປສາມາດໄດ້ຮັບການຢັ້ງຢືນໂດຍການທົດລອງຊ້ໍາກັນ.
2. Gene ID, ທ່ານຮູ້ຈັກ.
3. ຄວາມຍາວຂອງ gene, ຄວາມຍາວທັງຫມົດຂອງ gene ເປົ້າຫມາຍແມ່ນແນ່ນອນບໍ່ມີບັນຫາ.ເມື່ອອອກແບບ primers, ໃຫ້ແນ່ໃຈວ່າຄວາມຍາວຂອງ amplicon ແມ່ນຢູ່ລະຫວ່າງ 80-200bp ເພື່ອຮັບປະກັນປະສິດທິພາບການຂະຫຍາຍທີ່ດີຂຶ້ນ.
4. Sequence Blast ຂໍ້ມູນການປຽບທຽບ, gene ເປົ້າຫມາຍຕ້ອງໄດ້ຮັບການປຽບທຽບໃນ genebank ເພື່ອປ້ອງກັນບໍ່ໃຫ້ amplification ທີ່ບໍ່ແມ່ນສະເພາະ.
5. ການປະກົດຕົວຂອງ pseugenes.pseudogene ແມ່ນລໍາດັບ DNA ທີ່ຄ້າຍຄືກັບ gene ປົກກະຕິແຕ່ສູນເສຍຫນ້າທີ່ປົກກະຕິຂອງມັນ.ມັນມັກຈະມີຢູ່ໃນຄອບຄົວຫຼາຍເຊື້ອສາຍຂອງ eukaryotes.ປົກກະຕິແລ້ວມັນຖືກສະແດງໂດຍ ψ.ມັນແມ່ນການຄັດລອກ DNA genomic ທີ່ບໍ່ມີປະໂຫຍດໃນ genome ທີ່ຄ້າຍຄືກັບລໍາດັບ gene coding.ໂດຍທົ່ວໄປແລ້ວບໍ່ໄດ້ຖືກຖ່າຍທອດ, ແລະບໍ່ມີຄວາມຫມາຍທາງຊີວະວິທະຍາທີ່ຈະແຈ້ງ.
6. ຕຳແໜ່ງຂອງ primers ທຽບກັບ exons ແລະ introns.ໃນຊ່ວງຕົ້ນໆ, ເມື່ອພວກເຮົາແກ້ໄຂບັນຫາການປົນເປື້ອນຂອງ DNA, ພວກເຮົາມັກຈະເອົາໃຈໃສ່ກັບຕໍາແຫນ່ງຂອງ primers, exons, ແລະ introns, ແລະໂດຍທົ່ວໄປແລ້ວພິຈາລະນາການອອກແບບ primers ໃນທົ່ວ introns ເພື່ອຫຼີກເວັ້ນການຂະຫຍາຍ DNA.ກະລຸນາເບິ່ງຮູບຂ້າງລຸ່ມນີ້: ສີດໍາເປັນຕົວແທນຂອງ introns, blues ຕ່າງໆເປັນຕົວແທນຂອງ exons, ສີບົວເປັນຕົວແທນຂອງ primers ທົ່ວໄປ, ແລະສີແດງສົດໃສເປັນຕົວແທນຂອງ primers ພາຍໃນ.
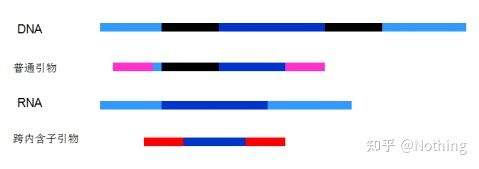
Schematic, ບໍ່ເຄີຍເປັນຄວາມຈິງ
ສິ່ງທີ່ເປັນແຜນການທີ່ສົມບູນແບບນີ້ເບິ່ງຄືວ່າ, ແຕ່ໃນຄວາມເປັນຈິງ, ໃນກໍລະນີຫຼາຍທີ່ສຸດ, primers trans-intron ແມ່ນບໍ່ magical ເທົ່າທີ່ຈິນຕະນາການ, ແລະພວກມັນຍັງຈະເຮັດໃຫ້ເກີດການຂະຫຍາຍທີ່ບໍ່ສະເພາະ.ດັ່ງນັ້ນວິທີທີ່ດີທີ່ສຸດເພື່ອປ້ອງກັນການປົນເປື້ອນຂອງ DNA ແມ່ນການກໍາຈັດ DNA ຢ່າງສົມບູນ.
7. ການຄາດຄະເນການສອດຄ່ອງ.ການນໍາໃຊ້ຕົວຢ່າງນີ້ອີກເທື່ອຫນຶ່ງ, ໃຊ້ເຄື່ອງມືອອນໄລນ໌ mFold ເພື່ອກໍານົດໂຄງສ້າງຂັ້ນສອງຂອງຊິ້ນເປົ້າຫມາຍຢູ່ທີ່ອຸນຫະພູມສະເພາະແລະຄວາມເຂັ້ມຂົ້ນຂອງເກືອ.
ໂຄງສ້າງຂັ້ນສອງຂອງ RNA ໃນອຸນຫະພູມທີ່ແຕກຕ່າງກັນ
ໂຄງສ້າງຂັ້ນສອງແມ່ນການຈັບຄູ່ແບບປະສົມປະສານຂອງແມ່ແບບຕົວມັນເອງເຊິ່ງຈະນໍາໄປສູ່ການແຂ່ງຂັນທີ່ເຂັ້ມແຂງລະຫວ່າງການຈັບຄູ່ primer ແລະ template, ແລະໂອກາດຂອງການຜູກມັດ primer ແມ່ນຫນ້ອຍ, ເຊິ່ງກໍ່ໃຫ້ເກີດບັນຫາຕ່າງໆເຊັ່ນ E ຕ່ໍາແລະການເຮັດເລື້ມຄືນທີ່ບໍ່ດີ.ຜ່ານການຄາດຄະເນຂອງຊອບແວ, ຖ້າບໍ່ມີບັນຫາໂຄງສ້າງຮອງ, ມັນຈະດີຫຼາຍ.ຖ້າມີ, ບົດຄວາມຕິດຕາມຂອງພວກເຮົາຈະສົນທະນາໂດຍສະເພາະວິທີການແກ້ໄຂບັນຫານີ້.
MIQE (6)—qPCR Oligonucleotides

ສໍາລັບ PCR ປະລິມານ fluorescent, ສິ່ງທໍາອິດທີ່ທ່ານຕໍ່ສູ້ກັບທຸກໆມື້ແມ່ນການສະກັດເອົາ RNA, ແລະສິ່ງທີສອງອາດຈະເປັນການອອກແບບ primer.
ກ່ອນອື່ນ ໝົດ, ພວກເຮົາຍັງກວດເບິ່ງກົດລະບຽບກ່ຽວກັບການອອກແບບ primer ຕາມລາຍການກວດກາ MIQE.ມັນງ່າຍດາຍຫຼາຍທີ່ຄົນຂີ້ຄ້ານສາມາດຫົວ, ແລະພວກເຮົາສາມາດເຮັດສໍາເລັດມັນໃນປະໂຫຍກຫນຶ່ງ: ຊອກຫາລໍາດັບແລະຕໍາແຫນ່ງຂອງ primer probe ແລະວິທີການດັດແປງ.ສໍາລັບວິທີການທໍາຄວາມສະອາດ primer, ການສັງເຄາະ primer ແມ່ນມີລາຄາຖືກຫຼາຍໃນປັດຈຸບັນ, qPCR ແມ່ນສົມຄວນກັບ PAGE ແລະຂ້າງເທິງວິທີການທໍາຄວາມສະອາດ, ແລະຂໍ້ມູນຂອງເຄື່ອງມືການສັງເຄາະແມ່ນບໍ່ສໍາຄັນ.ປະຊາຊົນຈໍານວນຫຼາຍໄດ້ເຮັດ primers ສໍາລັບທົດສະວັດແລະບໍ່ຮູ້ວ່າ synthesizer ແມ່ນ ABI3900.
ກ່ຽວກັບຫຼັກການຂອງການອອກແບບ primer, ທ່ານບໍ່ຈໍາເປັນຕ້ອງຈື່ໃຫ້ເຂົາເຈົ້າໂດຍ rote, ເພາະວ່າຊອບແວການອອກແບບ primer ສ່ວນໃຫຍ່ຫຼືເຄື່ອງມືອອນໄລນ໌ສາມາດດູແລບັນຫາເຫຼົ່ານີ້ໄດ້ (ແນະນໍາເຄື່ອງມືອອນໄລນ໌ primer3.ut.ee/), ແລະ 99.999% ຂອງການອອກແບບ primer ບໍ່ໄດ້ເຮັດດ້ວຍຕົນເອງເບິ່ງ, ຜູ້ຂຽນບາງຄັ້ງການອອກແບບຫຼາຍຮ້ອຍ primers ຕໍ່ມື້, ຖ້າຫາກວ່າທ່ານອ່ານຫນຶ່ງໂດຍຫນຶ່ງ, ມັນຈະກາຍເປັນຂ້າມ.
ພຽງແຕ່ກວດເບິ່ງຈຸດຕໍ່ໄປນີ້ຫຼັງຈາກ primers ຖືກອອກແບບມາ:
1. ການອອກແບບ primers ຢູ່ໃກ້ກັບປາຍ 3′: ໃນກໍລະນີຂອງການນໍາໃຊ້ primers oligo dT ສໍາລັບການສັງເຄາະສາຍທໍາອິດ cDNA, ພິຈາລະນາປະສິດທິພາບການຖອດຂໍ້ຄວາມແບບປີ້ນກັບກັນແລະຄວາມສົມບູນຂອງ RNA, primers ທີ່ຖືກອອກແບບຈໍາເປັນຕ້ອງໄດ້ອອກແບບຢູ່ໃກ້ກັບ 3′ ທີ່ສຸດເພື່ອປັບປຸງປະສິດທິພາບການຂະຫຍາຍ.ໃຊ້ຮູບເພື່ອອະທິບາຍດັ່ງຕໍ່ໄປນີ້ (ບໍ່ມີທາງທີ່ຈະເຂົ້າໃຈເລື່ອງນີ້):
ເປັນຫຍັງ primers ຄວນຖືກອອກແບບຢູ່ໃກ້ກັບ 3′, ມັນຈະບໍ່ເປັນຄວາມຈິງ
2. ຄ່າ TM: ຄ່າ Tm ຢູ່ທີ່ 55-65°C (ເນື່ອງຈາກວ່າກິດຈະກໍາ exonuclease ສູງທີ່ສຸດຢູ່ທີ່ 60°C), ແລະເນື້ອໃນ GC ຢູ່ທີ່ 40%-60%.
3. BLAST: ເພື່ອຫຼີກເວັ້ນການຂະຫຍາຍພັນທຸກໍາທີ່ບໍ່ສະເພາະ, Blast ຕ້ອງຖືກໃຊ້ສໍາລັບການຢັ້ງຢືນເພີ່ມເຕີມ.
MIQE(7)—ຂະບວນການ qPCR
1. ຊຸດ qPCR
ອີງຕາມຄວາມຕ້ອງການຂອງ MIQE, ພວກເຮົາຕ້ອງອະທິບາຍຢ່າງຈະແຈ້ງກ່ຽວກັບເງື່ອນໄຂການຕິກິຣິຍາທີ່ສົມບູນໃນບົດຄວາມ, ລວມທັງການຕັ້ງຄ່າຂອງລະບົບຕິກິຣິຍາ PCR, ຊຸດໃດທີ່ໃຊ້, ໃຜເປັນຜູ້ຜະລິດ, ລະບົບຕິກິຣິຍາໃຫຍ່ເທົ່າໃດ, ບໍ່ວ່າຈະໃຊ້ວິທີການຍ້ອມສີຫຼືວິທີການ probe, ການຕັ້ງຄ່າໂຄງການ PCR.ຜູ້ຂັບຂີ່ນັກຮົບເກົ່າຈະພົບວ່າຕາບໃດທີ່ຊຸດໄດ້ຖືກເລືອກ, ຂໍ້ມູນຂ້າງເທິງແມ່ນຖືກກໍານົດໂດຍພື້ນຖານ.
ໃນປັດຈຸບັນ, ການຜະລິດແລະການຜະລິດຊຸດ PCR ປະລິມານ fluorescent ແມ່ນເຕັກໂນໂລຢີທີ່ແກ່ຫຼາຍ.ຕາບໃດທີ່ທ່ານບໍ່ເລືອກຜູ້ຜະລິດທີ່ບໍ່ດີທີ່ສຸດ, ຄວາມເປັນໄປໄດ້ຂອງບັນຫາແມ່ນບໍ່ສູງ, ແຕ່ພວກເຮົາຍັງຕ້ອງການແບ່ງປັນບາງຈຸດກັບທ່ານ:
ເອນໄຊ Taq ເລີ່ມຕົ້ນຮ້ອນ:ສ່ວນທີ່ສໍາຄັນທີ່ສຸດຂອງ PCR ແມ່ນ enzyme Taq ເລີ່ມຕົ້ນຮ້ອນ.enzymes ເລີ່ມຕົ້ນຮ້ອນຢູ່ໃນຕະຫຼາດໂດຍທົ່ວໄປແມ່ນແບ່ງອອກເປັນສອງປະເພດ, ຫນຶ່ງແມ່ນ enzymes ເລີ່ມຕົ້ນຮ້ອນທີ່ຖືກດັດແປງທາງເຄມີ (ທ່ານສາມາດຈິນຕະນາການວ່າມັນເປັນການຝັງຕົວຂອງ paraffin), ແລະອີກອັນຫນຶ່ງແມ່ນ enzymes ເລີ່ມຕົ້ນຮ້ອນສໍາລັບການດັດແປງພູມຕ້ານທານ (antigen-antibody binding).ການປ່ຽນແປງທາງເຄມີແມ່ນວິທີການເລີ່ມຕົ້ນຂອງ enzymes ຮ້ອນ.ໃນເວລາທີ່ອຸນຫະພູມສະເພາະໃດຫນຶ່ງແມ່ນບັນລຸໄດ້, enzyme ຈະປ່ອຍກິດຈະກໍາຂອງຕົນ.ເອນໄຊເລີ່ມຮ້ອນທີ່ແກ້ໄຂດ້ວຍພູມຕ້ານທານໃຊ້ວິທີທາງຊີວະວິທະຍາເພື່ອສະກັດກັ້ນການເຄື່ອນໄຫວຂອງເອນໄຊ.ໃນເວລາທີ່ອຸນຫະພູມສະເພາະໃດຫນຶ່ງແມ່ນບັນລຸໄດ້, ພູມຕ້ານທານຈະໄດ້ຮັບການ denatured ແລະ inactivated ເປັນທາດໂປຼຕີນ, ແລະກິດຈະກໍາຂອງ enzyme ຈະຖືກນໍາເຂົ້າໄປໃນການຫຼິ້ນ.
ຢ່າງໃດກໍຕາມ, ການນໍາໃຊ້ນີ້ແມ່ນຫຍັງ?ນີ້ແມ່ນກໍລະນີ, ກິດຈະກໍາການປ່ອຍຕົວຂອງ enzymes ແກ້ໄຂພູມຕ້ານທານແມ່ນໄວກວ່າ enzymes ທີ່ຖືກດັດແປງທາງເຄມີ, ດັ່ງນັ້ນໃນແງ່ຂອງຄວາມອ່ອນໄຫວ, enzymes ແກ້ໄຂ antibody ມີປະໂຫຍດເລັກນ້ອຍ, ດັ່ງນັ້ນບໍ່ມີ enzymes ແກ້ໄຂທາງເຄມີໂດຍພື້ນຖານຢູ່ໃນຊຸດໃນຕະຫຼາດ.ຖ້າມີ, ເຕັກໂນໂລຢີຂອງຜູ້ຜະລິດນີ້ຍັງຕິດຢູ່ໃນຍຸກສະຫັດສະຫວັດ.
ຄວາມເຂັ້ມຂຸ້ນຂອງ magnesium ion:ຄວາມເຂັ້ມຂົ້ນຂອງ magnesium ion ມີຄວາມສໍາຄັນຫຼາຍໃນປະຕິກິລິຍາ PCR.ຄວາມເຂັ້ມຂົ້ນຂອງ magnesium ion ທີ່ເຫມາະສົມສາມາດສົ່ງເສີມການປ່ອຍກິດຈະກໍາ enzyme Taq.ຖ້າຄວາມເຂັ້ມຂົ້ນຕໍ່າເກີນໄປ, ກິດຈະກໍາຂອງ enzyme ຈະຫຼຸດລົງຢ່າງຫຼວງຫຼາຍ;ຖ້າຄວາມເຂັ້ມຂົ້ນສູງເກີນໄປ, ການຂະຫຍາຍຕົວທີ່ບໍ່ສະເພາະຂອງ enzyme-catalyzed ຈະຖືກປັບປຸງ.ຄວາມເຂັ້ມຂົ້ນຂອງ magnesium ions ຍັງຈະສົ່ງຜົນກະທົບຕໍ່ການຫມູນວຽນຂອງ primers, ອຸນຫະພູມ melting ຂອງແມ່ແບບແລະຜະລິດຕະພັນ PCR, ດັ່ງນັ້ນຜົນກະທົບຕໍ່ຜົນຜະລິດຂອງ fragments amplified.ຄວາມເຂັ້ມຂຸ້ນຂອງ magnesium ions ໂດຍທົ່ວໄປແມ່ນຖືກຄວບຄຸມຢູ່ທີ່ 25mM.ແນ່ນອນ, ສໍາລັບຊຸດທີ່ດີ, ຄວາມເຂັ້ມຂົ້ນຂອງ magnesium ions ຕ້ອງໄດ້ຮັບການຄວບຄຸມໄດ້ດີ.ພໍ່ຄ້າບາງຄົນເພີ່ມສານ magnesium ion chelating agent ໃຫ້ກັບ reagent, ເຊິ່ງສາມາດບັນລຸຜົນຂອງການປັບອັດຕະໂນມັດຂອງຄວາມເຂັ້ມຂົ້ນຂອງ magnesium ion.
ຄວາມເຂັ້ມຂຸ້ນຂອງສີຍ້ອມ fluorescent:ສີຍ້ອມ fluorescent, ເຊິ່ງເປັນ SYBR Green ທີ່ພວກເຮົາໃຊ້ເລື້ອຍໆ, ສ່ວນໃຫຍ່ແມ່ນສ້າງ fluorescence ໂດຍການຜູກມັດກັບຮ່ອງເລັກນ້ອຍຂອງ DNA ສອງສາຍ, ເພາະວ່າການຜູກມັດຂອງສີຍ້ອມກັບ DNA ເສັ້ນຄູ່ແມ່ນບໍ່ສະເພາະ, ນັ້ນແມ່ນ, ຕາບໃດທີ່ DNA ຄູ່ສາຍຖືກລວມເຂົ້າກັນກັບມັນ, fluorescence ຈະເກີດຂື້ນໃນພື້ນຖານຂອງລະບົບ DNA. ສັນຍານ.
PS: ເນື່ອງຈາກຄຸນສົມບັດທີ່ມີຄວາມອ່ອນໄຫວຕໍ່ແສງສະຫວ່າງຂອງມັນ, ຜະລິດຕະພັນຢູ່ໃນຕະຫຼາດໂດຍທົ່ວໄປໄດ້ຖືກຫຸ້ມຫໍ່ຢູ່ໃນທໍ່ centrifuge ສີນ້ໍາຕານ opaque (ດັ່ງທີ່ສະແດງຢູ່ໃນຮູບຂ້າງລຸ່ມນີ້).ຢ່າງໃດກໍຕາມ, ນີ້ຈະພົບກັບບັນຫາ.ມັນເປັນການຍາກທີ່ຈະເບິ່ງວ່າຂອງແຫຼວຖືກດູດໃນເວລາເກັບຕົວຢ່າງ.ໃນເລື່ອງນີ້, Qingke ແມ່ນແທ້ໆທີ່ເປັນມິດກັບຜູ້ໃຊ້ທີ່ສຸດ (ດັ່ງທີ່ສະແດງຢູ່ໃນຮູບຂ້າງລຸ່ມນີ້), ແລະທໍ່ໂປ່ງໃສຖືກຫຸ້ມຫໍ່ຢູ່ໃນຖົງກົ່ວ opaque.ຫຼັງຈາກນັ້ນ, ເອົາມັນເຂົ້າໄປໃນຖົງກົ່ວ, ຄໍານຶງເຖິງຄວາມສະດວກສະບາຍຂອງການຫຼີກເວັ້ນການແສງສະຫວ່າງແລະການເກັບຕົວຢ່າງ.ທ່ານຕ້ອງເລືອກຈໍານວນຜະລິດຕະພັນທີ່ຖືກຕ້ອງ.TSE204 ເປັນສິ່ງມີຊີວິດທີ່ຄຸ້ມຄ່າທີ່ສຸດ, ເຊິ່ງເຮັດໃຫ້ຂ້ອຍຢາກປູກຫຍ້າ.

ຄວາມເຂັ້ມຂຸ້ນຂອງສີຍ້ອມ fluorescent ຍັງມີຄວາມສໍາຄັນຫຼາຍ.ຖ້າຄວາມເຂັ້ມຂົ້ນຕ່ໍາເກີນໄປ, ເສັ້ນໂຄ້ງຂະຫຍາຍຈະບໍ່ຂຶ້ນໃນຂັ້ນຕອນຕໍ່ມາແລະບໍ່ສົມບູນແບບ;ຖ້າຄວາມເຂັ້ມຂຸ້ນສູງເກີນໄປ, ມັນຈະເຮັດໃຫ້ເກີດການລົບກວນສິ່ງລົບກວນ.ເນື່ອງຈາກ PCR ປະລິມານ fluorescent ສ່ວນໃຫຍ່ແມ່ນຂຶ້ນກັບຄ່າ CT, ຖ້າຄວາມເຂັ້ມຂົ້ນຂອງສີຍ້ອມ fluorescent ບໍ່ໄດ້ຖືກປັບຢ່າງຖືກຕ້ອງ, ຈຸດຕ່ໍາແມ່ນດີກ່ວາຈຸດສູງ.ແນ່ນອນ, ຄວາມເຂັ້ມຂົ້ນຂອງສີຍ້ອມທີ່ເຫມາະສົມແມ່ນດີທີ່ສຸດ.
ROX: ສີຍ້ອມ ROX ຖືກນໍາໃຊ້ເພື່ອແກ້ໄຂຄວາມຜິດພາດຂອງສັນຍານ fluorescence ທີ່ດີ.ບາງຜູ້ຜະລິດເຄື່ອງມືຮຽກຮ້ອງໃຫ້ມີການປັບທຽບ, ໃນຂະນະທີ່ຄົນອື່ນບໍ່ເຮັດ.ຕົວຢ່າງເຊັ່ນ, ການນໍາໃຊ້ເຄື່ອງມືຂະຫຍາຍ PCR ເວລາຈິງຂອງ Thermo Fisher Scientific ປົກກະຕິແລ້ວຮຽກຮ້ອງໃຫ້ມີການປັບຕົວ, ລວມທັງ 7300, 7500, 7500Fast, StepOnePlus, ແລະອື່ນໆ ຄໍາແນະນໍາຊຸດທົ່ວໄປຈະອະທິບາຍມັນ.
qPCR Mix ຂອງ Foregene ຍັງມີສີຍ້ອມ ROX, ເຊິ່ງສະດວກໃນການນໍາໃຊ້ໃນແບບຕ່າງໆ.
ການປິ່ນປົວພັນທະບັດ hydrogen ອ່ອນແອ: ການປິ່ນປົວພັນທະບັດໄຮໂດເຈນທີ່ອ່ອນແອແມ່ນເປັນເລື່ອງດ້ານວິຊາການທີ່ຂ້ອນຂ້າງ.ບໍ່ມີຫຍັງໄດ້ອ່ານຄູ່ມືຂອງຊຸດຫຼາຍ, ແຕ່ບໍ່ມີໃຜໄດ້ກ່າວເຖິງຫົວຂໍ້ນີ້.ໃນຄວາມເປັນຈິງ, ມັນເປັນສິ່ງສໍາຄັນຫຼາຍ.ການປະສົມປະສານຂອງພື້ນຖານສ່ວນໃຫຍ່ແມ່ນຂຶ້ນກັບຄວາມເຂັ້ມແຂງຂອງພັນທະບັດ hydrogen.ພັນທະບັດໄຮໂດເຈນທີ່ເຂັ້ມແຂງແມ່ນການຂະຫຍາຍປົກກະຕິ, ແລະພັນທະບັດໄຮໂດເຈນທີ່ອ່ອນແອນໍາໄປສູ່ການຂະຫຍາຍທີ່ບໍ່ສະເພາະ.ຖ້າພັນທະບັດໄຮໂດເຈນທີ່ອ່ອນແອບໍ່ສາມາດຖືກກໍາຈັດໄດ້ດີ, ການຂະຫຍາຍທີ່ບໍ່ສະເພາະແມ່ນບໍ່ສາມາດຫຼີກເວັ້ນໄດ້.ພາຍໃນຂອບເຂດຂອງຜູ້ຂຽນ, ມີພຽງແຕ່ບໍລິສັດຈໍານວນຫນ້ອຍທີ່ສັງເກດເຫັນບັນຫານີ້.ເມື່ອທ່ານຊື້ຊຸດ, ທ່ານສາມາດອ້າງອີງເຖິງວ່າທ່ານໄດ້ພິຈາລະນາການແກ້ໄຂໃນເລື່ອງນີ້ສໍາລັບຊຸດທີ່ທ່ານຕ້ອງການທີ່ຈະເລືອກເອົາ.
ປະລິມານປະຕິກິລິຍາ: ລະບົບ 20-50ul ແມ່ນໃຊ້ທົ່ວໄປຫຼາຍ, ແລະປະລິມານຂະຫນາດນ້ອຍກວ່າຈະເຮັດໃຫ້ເກີດຄວາມຜິດພາດ.ໂດຍທົ່ວໄປແລ້ວ, ຄໍາແນະນໍາຊຸດຈະແນະນໍາໃຫ້ໃຊ້ປະລິມານຕິກິຣິຍາ PCR.ຢ່າສະຫຼາດ ແລະໃຊ້ປະລິມານໜ້ອຍລົງເພື່ອປະຢັດຄ່າໃຊ້ຈ່າຍ.ເປົ້າຫມາຍຂອງ.ປະລິມານທີ່ແນະນໍາໂດຍຜູ້ຄ້າໄດ້ຮັບການທົດສອບຕົວຈິງແລ້ວ, ແລະມັນອາດຈະວ່າພວກເຂົາບໍ່ສາມາດແກ້ໄຂບັນຫາຄວາມຜິດພາດທີ່ເກີດຈາກປະລິມານຂະຫນາດນ້ອຍ.
2. ຜູ້ຜະລິດແລະຈໍານວນບົດຄວາມຂອງແຜ່ນທໍ່
ທຸກຄົນຮູ້ຫຼັກການຂອງ PCR ປະລິມານ fluorescent.ການເກັບກໍາ fluorescence ສ່ວນໃຫຍ່ແມ່ນດໍາເນີນໂດຍຜ່ານຫມວກທໍ່ PCR.ເມື່ອເລືອກເຄື່ອງບໍລິໂພກ PCR, ໃຫ້ເອົາໃຈໃສ່ສອງຈຸດ: ການສົ່ງແສງສະຫວ່າງທີ່ດີແລະເຫມາະສົມກັບເຄື່ອງມື.ໂດຍທົ່ວໄປແລ້ວ, ກະດານແລະທໍ່ຂອງຍີ່ຫໍ້ຕົ້ນຕໍແມ່ນດີ, ແຕ່ທ່ານຕ້ອງເລືອກຢ່າງລະມັດລະວັງໃນການປັບຕົວ, ຖ້າບໍ່ດັ່ງນັ້ນທ່ານຈະບໍ່ສາມາດໃຊ້ເຄື່ອງມື.

4. ຄວາມຮູ້ລະດັບສູງສຸດ
MIQE (8)—ການກວດສອບ qPCR
ນີ້ແມ່ນບູລິມະສິດສູງສຸດຂອງ qPCR!ດັ່ງນັ້ນວິລະຊົນຈໍານວນຫຼາຍໄດ້ຕົກເຂົ້າໄປໃນດິນຊາຍຢູ່ທີ່ນີ້.ແນ່ນອນ, ມັນກໍ່ເປັນໄປໄດ້ວ່າເຈົ້າໂຊກດີແລະພັນທຸກໍາທີ່ເຈົ້າສຶກສາແມ່ນງ່າຍດາຍ, ດັ່ງນັ້ນເຈົ້າຈຶ່ງລອຍຕົວຜ່ານຖ້ໍາກ້ອນຕາມສາຍລົມ.ຂໍ້ມູນການກວດສອບຂອງ qPCR ມີຈຸດປະສົງເພື່ອທົດສອບຄວາມຫນ້າເຊື່ອຖືຂອງຂໍ້ມູນ.ພວກເຮົາລະບຸຂໍ້ມູນການກວດສອບທີ່ຈໍາເປັນດັ່ງນີ້:
1.ການທົດສອບສະເພາະ
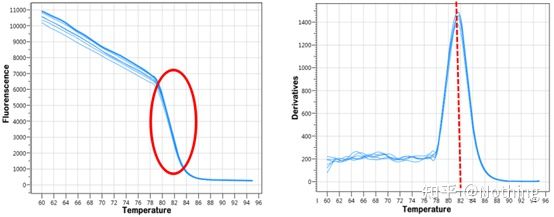
ຄວາມສະເພາະຂອງການຂະຫຍາຍພັນທຸກໍາເປົ້າຫມາຍແມ່ນການທົດສອບໂດຍການກວດສອບວ່າຮູບພາບ electrophoresis ເປັນແຖບດຽວ;ການກວດສອບລໍາດັບ;melting curve ເພື່ອເບິ່ງວ່າແຜນທີ່ສູງສຸດແມ່ນອັນດຽວ;ການກວດສອບການຍ່ອຍອາຫານຂອງ enzyme ແລະວິທີການອື່ນໆ.
ທີ່ນີ້, ພວກເຮົາສຸມໃສ່ tລາວວິເຄາະການຂະຫຍາຍທີ່ບໍ່ສະເພາະໂດຍວິທີການຂອງເສັ້ນໂຄ້ງ melting.ໂດຍທົ່ວໄປແລ້ວ, ເມື່ອພວກເຮົາອອກແບບ primers, ຂະຫນາດຂອງຊິ້ນຜະລິດຕະພັນແມ່ນຕ້ອງການຢູ່ໃນລະດັບ 80-200bp, ເຊິ່ງເຮັດໃຫ້ອຸນຫະພູມການລະລາຍຂອງຜະລິດຕະພັນ PCR ຢູ່ໃນລະດັບ 80-85 ° C.ດັ່ງນັ້ນ, ຖ້າມີຈຸດສູງສຸດອື່ນໆ, ຕ້ອງມີຜະລິດຕະພັນຂະຫຍາຍອື່ນໆທີ່ບໍ່ສະເພາະ;ຖ້າຈຸດສູງສຸດປາກົດຢູ່ຕ່ໍາກວ່າ 80 ° C, ໂດຍທົ່ວໄປແລ້ວມັນຖືກພິຈາລະນາວ່າເປັນ primer dimer;ຖ້າຈຸດສູງສຸດປາກົດຢູ່ຂ້າງເທິງ 85 ° C, ໂດຍທົ່ວໄປແລ້ວມັນຖືກພິຈາລະນາວ່າເປັນການປົນເປື້ອນຂອງ DNA ຫຼືການຂະຫຍາຍທີ່ບໍ່ສະເພາະຂອງຊິ້ນຂະຫນາດໃຫຍ່.
ໝາຍເຫດ: ບາງຄັ້ງມີພຽງຈຸດດຽວຢູ່ທີ່ 80°C.ໃນເວລານີ້, ແນວຄວາມຄິດນີ້ຕ້ອງໄດ້ຮັບການຍຶດຫມັ້ນ.ມັນເປັນໄປໄດ້ວ່າຜົນໄດ້ຮັບການຂະຫຍາຍແມ່ນ primer dimers ທັງຫມົດ.
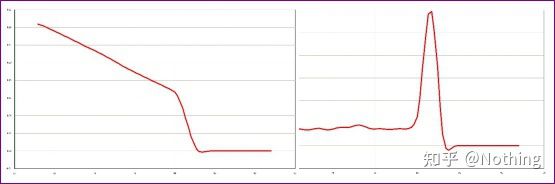
ເສັ້ນໂຄ້ງການລະລາຍປົກກະຕິ (ຈຸດສູງສຸດດຽວທີ່ບໍ່ມີການຂະຫຍາຍຕົວທີ່ບໍ່ແມ່ນສະເພາະ)
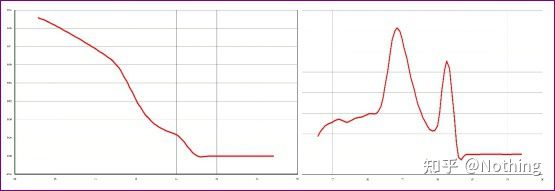
ເສັ້ນໂຄ້ງການລະລາຍທີ່ເປັນບັນຫາ (ການຂະຫຍາຍຕົວທີ່ບໍ່ແມ່ນສະເພາະຂອງຈຸດສູງສຸດ spurious)
【ການວິເຄາະກໍລະນີ】
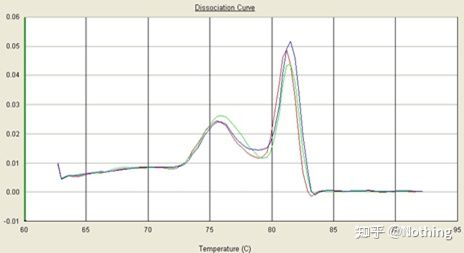
ມີຈຸດສູງສຸດຕົ້ນຕໍ, ແຕ່ primer dimer ແມ່ນຮ້າຍແຮງ
ເສັ້ນໂຄ້ງການລະລາຍຈຸດສູງສຸດດຽວໃນຮູບຂ້າງລຸ່ມນີ້ສາມາດຫລອກລວງຕາຂອງທ່ານໄດ້ຢ່າງງ່າຍດາຍ, ຄິດວ່າມັນເປັນການທົດລອງທີ່ສົມບູນແບບ, ແຕ່ຜົນໄດ້ຮັບແມ່ນຜິດພາດຫມົດ.ໃນເວລານີ້, ພວກເຮົາຕ້ອງເບິ່ງອຸນຫະພູມຂອງການລະລາຍ.ອຸນຫະພູມສູງສຸດແມ່ນຕໍ່າກວ່າ 80 ອົງສາ C, ເຊິ່ງເປັນ primer-dimer ຢ່າງສົມບູນ.
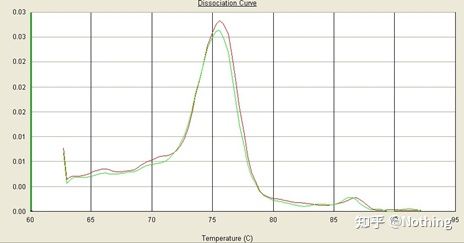
ບໍ່ມີຊິ້ນສ່ວນເປົ້າໝາຍ, ທັງໝົດ primer dimers
ນີ້, ອ້າຍຂອງຂ້ອຍບໍ່ສາມາດຢຸດໄດ້.ຮູບຂ້າງລຸ່ມນີ້ແມ່ນຮູບທີ່ຖ່າຍດ້ວຍໂທລະສັບມືຖືສົ່ງໃຫ້ຂ້ອຍໂດຍຄົນຂີ້ຄ້ານ.reagents ທີ່ເຂົາໃຊ້ແມ່ນທັງຫມົດຍີ່ຫໍ້ທີ່ໃຊ້ທົ່ວໄປໃນອຸດສາຫະກໍາ.ລາວໄດ້ປ່ຽນຈາກຍີ່ຫໍ້ T-prefix ໄປຫາຍີ່ຫໍ້ T-prefix ອື່ນ.ຂ້ອຍຄິດວ່າເຈົ້າໄດ້ເດົາແລ້ວ.ຄົນຂີ້ຄ້ານຮ້ອງໃສ່ຂ້ອຍວ່າ: “ນໍ້າຢາທີ່ໃຊ້ໃນຮູບທຳອິດແມ່ນດີເກີນໄປ, ແລະຈຸດສູງສຸດແມ່ນອັນດຽວ.ຕໍ່ມາ, ຫຼັງຈາກໃຊ້ reagent ທີ່ທ່ານແນະນໍາ, ມັນຈະກາຍເປັນຄືກັບຮູບທີສອງ, ມີຈຸດສູງສຸດປະສົມ.ເຈົ້າໄດ້ເຮັດໃຫ້ຂ້ອຍທຸກທໍລະມານ.“
ແຍກທັງສອງກາຟ.ຢູ່ glance ທໍາອິດ, ຫນຶ່ງມີຈຸດສູງສຸດດຽວ, ແລະອີກອັນຫນຶ່ງມີຈຸດສູງສຸດສອງເທົ່າ.ບໍ່ມີເຫດຜົນ, ສູງສຸດດຽວແມ່ນແນ່ນອນດີ.ແມ່ນຄວາມຈິງທີ່ວ່າ?
ຮ້າຍກວ່າ Dou E, ຖ້າຂ້ອຍໃສ່ສອງຮູບຂ້າງລຸ່ມນີ້, ເຈົ້າຈະເຂົ້າໃຈທັນທີ.ໃນຄວາມເປັນຈິງ, ພວກເຮົາເປັນອໍາມະພາດໄດ້ຢ່າງງ່າຍດາຍໂດຍຮູບແບບນີ້.ຫຼັງຈາກການວິເຄາະລະມັດລະວັງ, ພວກເຮົາໄດ້ພົບເຫັນວ່າ: ຈຸດສູງສຸດຂອງຕົວເລກທໍາອິດແມ່ນຢູ່ທີ່ 75°C, ເຊິ່ງແມ່ນ primer dimer ຫມົດ;ຈຸດສູງສຸດຂອງຕົວເລກທີສອງປາກົດຢູ່ທີ່ 75 ° C ແລະ 82 ° C, ຢ່າງຫນ້ອຍມີຜະລິດຕະພັນປະກົດວ່າ.
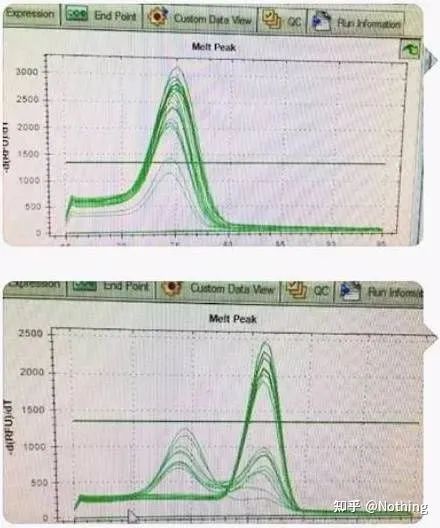
ຮູບພາບການຕິຊົມຈາກນັກຮຽນ
ດັ່ງນັ້ນບັນຫາພື້ນຖານບໍ່ແມ່ນບັນຫາຂອງ reagents, ແຕ່ບັນຫາຂອງການອອກແບບ primer.ໃນເວລາດຽວກັນ, ມັນຍັງພິສູດວ່າບາງຍີ່ຫໍ້ໃຫຍ່ບໍ່ມີຄຸນນະພາບຂອງທາດເຫຼັກ, ແລະມັນຍັງພິສູດສິ່ງທີ່ອ້າຍຂອງຂ້ອຍເວົ້າກ່ອນຫນ້ານີ້: ມັນບໍ່ແມ່ນຍີ່ຫໍ້ reagent ທີ່ສະຫນັບສະຫນູນບົດຄວາມຂອງເຈົ້າ.ມັນແມ່ນບົດຄວາມຂອງເຈົ້າທີ່ສົ່ງເສີມຍີ່ຫໍ້ຂອງ reagents.ພຽງແຕ່ຈິນຕະນາການ, ຖ້າ scumbag ບໍ່ໄດ້ປ່ຽນທາດ reagents, ຂໍ້ມູນທີ່ບໍ່ຖືກຕ້ອງຈະຖືກສົ່ງກັບວາລະສານ, ແລະສິ່ງທີ່ຈະເກີດຂື້ນຈະເປັນຄວາມໂສກເສົ້າ.
2. ຄ່າ Ct ຂອງການຄວບຄຸມເປົ່າ
ຢ່າອະທິບາຍ, ຖ້າການຄວບຄຸມເປົ່າມີຄ່າ Ct, ບໍ່ແມ່ນມົນລະພິດບໍ?ຢ່າງໃດກໍຕາມ, ທ່ານຍັງຈໍາເປັນຕ້ອງເຂົ້າໃຈວ່າການຄວບຄຸມເປົ່າໃດມີມູນຄ່າ Ct.ຖ້າມັນເປັນ NTC, ມັນຫມາຍຄວາມວ່າມີ DNA ຕ່າງປະເທດເຊັ່ນການປົນເປື້ອນ reagent.ຖ້າມັນເປັນ NRT, ມັນຫມາຍຄວາມວ່າ RNA ທີ່ສະກັດອອກມາມີການປົນເປື້ອນ DNA.
3. ເສັ້ນໂຄ້ງມາດຕະຖານ
ລວມທັງຄວາມຊັນແລະສູດການຄິດໄລ່, ປະສິດທິພາບ PCR ສາມາດຄິດໄລ່ໄດ້ໂດຍຜ່ານສູດ.ການທົດລອງທີ່ສົມບູນແບບຕ້ອງການຄວາມຊັນຂອງເສັ້ນໂຄ້ງມາດຕະຖານເພື່ອເຂົ້າຫາ 3.32, ແລະ R² ເຂົ້າຫາ 0.9999.
4. Linear dynamic range
ລະດັບການເຄື່ອນໄຫວຂອງປະຕິກິລິຍາແມ່ນເສັ້ນຊື່.ອີງຕາມແມ່ແບບທີ່ໃຊ້ເພື່ອສ້າງເສັ້ນໂຄ້ງມາດຕະຖານ, ຊ່ວງໄດນາມິກຄວນປະກອບມີ gradients ຄວາມເຂັ້ມຂົ້ນຢ່າງຫນ້ອຍ 5, ແລະເອົາໃຈໃສ່ກັບການປ່ຽນແປງຂອງຄ່າ Ct ໃນລະດັບຄວາມເຂັ້ມຂົ້ນສູງແລະ gradients ຄວາມເຂັ້ມຂົ້ນຕ່ໍາ.
5. ການກວດສອບຄວາມຖືກຕ້ອງ
ການປ່ຽນແປງໃນຜົນໄດ້ຮັບ qPCR, ນັ້ນແມ່ນ, ການເຮັດຊ້ໍາຄືນທີ່ບໍ່ດີ, ນັ້ນແມ່ນ, ຄວາມແມ່ນຍໍາທີ່ບໍ່ດີ, ແມ່ນເກີດມາຈາກຫຼາຍປັດໃຈ, ລວມທັງອຸນຫະພູມ, ຄວາມເຂັ້ມຂົ້ນ, ແລະການດໍາເນີນງານ.qPCR ຄວາມແມ່ນຍໍາໂດຍທົ່ວໄປຈະກາຍເປັນການຄວບຄຸມຫນ້ອຍລົງຍ້ອນວ່າຈໍານວນສໍາເນົາຫຼຸດລົງ.ການປ່ຽນແປງພາຍໃນການທົດລອງທີ່ເຫມາະສົມ, ການປ່ຽນແປງທາງດ້ານເຕັກນິກນີ້ຄວນຈະແຕກຕ່າງຈາກການປ່ຽນແປງທາງຊີວະພາບ, ແລະການຈໍາລອງທາງຊີວະພາບສາມາດແກ້ໄຂຄວາມແຕກຕ່າງທາງສະຖິຕິໂດຍກົງໃນຜົນໄດ້ຮັບ qPCR ລະຫວ່າງກຸ່ມຫຼືການປິ່ນປົວ.ໂດຍສະເພາະສໍາລັບການວິເຄາະວິນິດໄສ, ຄວາມແມ່ນຍໍາລະຫວ່າງການທົດສອບທີ່ດີທີ່ສຸດ (ການເຮັດຊ້ໍາອີກຄັ້ງ) ໃນທົ່ວເວັບໄຊທ໌ແລະຜູ້ປະຕິບັດການຕ້ອງໄດ້ຮັບການລາຍງານ.
6. ປະສິດທິພາບການກວດສອບ ແລະ LOD (ໃນ multiplex qPCR)
LOD ແມ່ນຄວາມເຂັ້ມຂົ້ນຕ່ໍາສຸດຂອງ 95% ຂອງຕົວຢ່າງໃນທາງບວກທີ່ກວດພົບ.ໃນຄໍາສັບຕ່າງໆອື່ນໆ, ຄວາມເຂັ້ມຂົ້ນຂອງ LOD ທີ່ມີຢູ່ໃນຊຸດຂອງຕົວແບບ gene ເປົ້າຫມາຍບໍ່ຄວນເກີນ 5% ຂອງປະຕິກິລິຍາທີ່ລົ້ມເຫລວ.ໃນເວລາທີ່ເຮັດການວິເຄາະ multiplex qPCR, ໂດຍສະເພາະສໍາລັບການກວດພົບພ້ອມໆກັນຂອງການກາຍພັນຂອງຈຸດຫຼື polymorphisms, multiplex qPCR ຕ້ອງການໃຫ້ຫຼັກຖານວ່າຄວາມຖືກຕ້ອງຂອງຊິ້ນສ່ວນເປົ້າຫມາຍຫຼາຍບໍ່ໄດ້ຖືກຫຼຸດຫນ້ອຍລົງໃນທໍ່ດຽວກັນ, ການກວດສອບຫຼາຍແລະການກວດພົບທໍ່ດຽວທີ່ມີປະສິດທິພາບແລະ LOD ຄວນຈະຄືກັນ.ໂດຍສະເພາະໃນເວລາທີ່ genes ເປົ້າຫມາຍທີ່ມີຄວາມເຂັ້ມຂຸ້ນສູງແລະ genes ເປົ້າຫມາຍທີ່ມີຄວາມເຂັ້ມຂຸ້ນຕ່ໍາໄດ້ຖືກຂະຫຍາຍໄປພ້ອມໆກັນ, ບັນຫານີ້ຕ້ອງໄດ້ຮັບການເອົາໃຈໃສ່.
ບັນຫາແລະວິທີແກ້ໄຂໂດຍທົ່ວໄປແລ້ວ, ບັນຫາທີ່ພົບເລື້ອຍໆໃນການດີບັກ qPCR ສຸມໃສ່ລັກສະນະດັ່ງຕໍ່ໄປນີ້:
·ການຂະຫຍາຍທີ່ບໍ່ສະເພາະ
·ທາງເລືອກທີ່ຍາກຂອງຄວາມເຂັ້ມຂົ້ນ primer ແລະບັນຫາກັບ primer-dimer
· ອຸນ ຫະ ພູມ annealing ແມ່ນ ບໍ່ ຖືກ ຕ້ອງ
·ໂຄງສ້າງຮອງມີຜົນກະທົບປະສິດທິພາບການຂະຫຍາຍ
ການຂະຫຍາຍທີ່ບໍ່ສະເພາະ
ການຂະຫຍາຍທີ່ບໍ່ສະເພາະເກີດຂື້ນ, ໂດຍທົ່ວໄປແລ້ວມັນຖືກພິຈາລະນາວ່າການອອກແບບ primer ແມ່ນບໍ່ເຫມາະສົມ, ແຕ່ຖ້າທ່ານບໍ່ຮີບຮ້ອນທີ່ຈະປ່ຽນ primers, ທ່ານສາມາດລອງວິທີຕໍ່ໄປນີ້ກ່ອນ (ຫຼັກການຍັງຕິດຢູ່):
·ເພີ່ມອຸນຫະພູມ annealing - ພະຍາຍາມເພື່ອເຮັດໃຫ້ພັນທະບັດ hydrogen ອ່ອນແອບໍ່ສາມາດຮັກສາ;
· ຫຼຸດການເຊື່ອມ ແລະເວລາການຍືດຕົວ - ຫຼຸດຜ່ອນໂອກາດຂອງພັນທະບັດໄຮໂດເຈນທີ່ອ່ອນແອ;
· ຫຼຸດຜ່ອນຄວາມເຂັ້ມຂຸ້ນ primer - ຫຼຸດຜ່ອນໂອກາດຂອງການຜູກມັດຂອງ primers redundant ແລະພາກພື້ນທີ່ບໍ່ແມ່ນເປົ້າຫມາຍ;
ປະສິດທິພາບການຂະຫຍາຍຕ່ໍາ
ສະຖານະການກົງກັນຂ້າມກັບການຂະຫຍາຍທີ່ບໍ່ສະເພາະ - ປະສິດທິພາບການຂະຫຍາຍຕ່ໍາ, ແລະມາດຕະການເພື່ອຈັດການກັບປະສິດທິພາບການຂະຫຍາຍຕ່ໍາແມ່ນກົງກັນຂ້າມ:
·ຍືດເວລາການຫມູນວຽນແລະຍືດຍາວ;
·ປ່ຽນເປັນສາມຂັ້ນຕອນ PCR ແລະຫຼຸດຜ່ອນອຸນຫະພູມ annealing;
·ເພີ່ມຄວາມເຂັ້ມຂົ້ນ primer;
Ps: ນັກສຶກສາຈົບການສຶກສາຈໍານວນຫຼາຍທີ່ເກີດໃນ 90s ບໍ່ເຕັມໃຈທີ່ຈະສຶກສາວິທີການ debug ທົດລອງ, ແລະຫວັງວ່າຊຸດສາມາດແກ້ໄຂບັນຫາໄດ້ຢ່າງສົມບູນ (ຖ້າຫາກວ່າທ່ານຕ້ອງການທີ່ຈະໄປບໍລິສັດ reagent ເພື່ອເຮັດການຄົ້ນຄວ້າແລະການພັດທະນາຫຼັງຈາກຮຽນຈົບ), ໃນຄວາມເປັນຈິງ, ຜູ້ຜະລິດ reagent ຍັງຄິດວິທີນີ້, ຂ້າພະເຈົ້າຫວັງວ່າມັນເປັນຄົນໂງ່ທີ່ມັນສາມາດໃຊ້ໄດ້ໃນເວລາທີ່ທ່ານໄດ້ຮັບມັນ, ດັ່ງນັ້ນຜູ້ຜະລິດ reagent ເພື່ອແກ້ໄຂບັນຫາທີ່ອ່ອນແອ, ລວມທັງຄວາມອ່ອນແອຂອງ H. - ປັດໄຈການດູດຊຶມພັນທະບັດ.ເພື່ອແກ້ໄຂບັນຫາໄດ້ງ່າຍ, ຄົນໂງ່ຍັງຕ້ອງອ່ານການແນະນໍາຂອງບໍລິສັດ reagent ເພື່ອເບິ່ງວ່າມີປັດໃຈທີ່ດູດຊຶມພັນທະບັດໄຮໂດເຈນທີ່ອ່ອນແອ.
ທາງເລືອກທີ່ຍາກຂອງຄວາມເຂັ້ມຂົ້ນ primer ແລະບັນຫາກັບ primer-dimer
ວິທີການ 1: ໂດຍທົ່ວໄປແລ້ວ, ຄໍາແນະນໍາຂອງຊຸດສໍາລັບ qPCR ມີລະບົບທີ່ແນະນໍາແລະຄວາມເຂັ້ມຂຸ້ນຂອງ primer ທີ່ແນະນໍາ.
ວິທີການ 2: ການດີບັກໂດຍການຕັ້ງລະດັບຄວາມເຂັ້ມຂຸ້ນຂອງ primer.ຮູບຂ້າງລຸ່ມນີ້ຖືກລັກຈາກບໍລິສັດເພື່ອສະແດງໃຫ້ເຫັນ.ຮູບຂ້າງລຸ່ມນີ້ສະແດງໃຫ້ເຫັນຜົນໄດ້ຮັບດ້ານປະລິມານ fluorescence ທີ່ເຮັດດ້ວຍສາມລະດັບຄວາມເຂັ້ມຂຸ້ນຂອງ primer (100nM, 250nM, 500nM) ແລະສີ່ gradients ຄວາມເຂັ້ມຂຸ້ນຂອງແມ່ແບບ (0.1ng, 1ng, 10ng, 100ng).ຄ່າ Ct ຂອງຜົນການທົດລອງແມ່ນໄດ້ວາງແຜນດັ່ງຕໍ່ໄປນີ້:
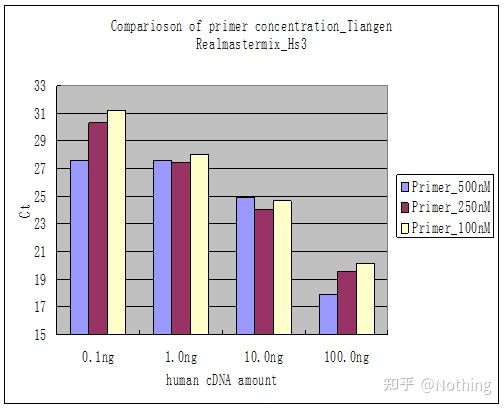
ການເລືອກຄວາມເຂັ້ມຂຸ້ນຂອງ primer ສົມທົບຄວາມເຂັ້ມຂຸ້ນຂອງ primer ແຕ່ລະອັນເປັນເສັ້ນດັ່ງຕໍ່ໄປນີ້:
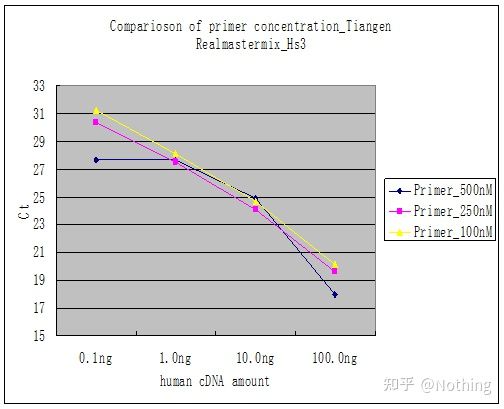
ທາງເລືອກຂອງຄວາມເຂັ້ມຂົ້ນຂອງ primer ແມ່ນເຫັນໄດ້ຊັດເຈນ, ຄວາມສໍາພັນເສັ້ນຂອງຄວາມເຂັ້ມຂົ້ນ primer ຂອງ 100nM ແລະ 250nM ແມ່ນດີກວ່າ, ແລະຄວາມສໍາພັນເສັ້ນຂອງຄວາມເຂັ້ມຂົ້ນ primer ຂອງ 500nM ແມ່ນຂ້ອນຂ້າງບໍ່ດີ.ໃນ 100nM ແລະ 250nM, ຄ່າ Ct ຂອງ 250nM ແມ່ນຂ້ອນຂ້າງນ້ອຍ, ດັ່ງນັ້ນຄວາມເຂັ້ມຂຸ້ນຂອງ primer ທີ່ດີທີ່ສຸດແມ່ນ 250nM.ໂດຍທົ່ວໄປແລ້ວ primer-dimers ຮ້າຍແຮງສາມາດເຫັນໄດ້ໃນເສັ້ນໂຄ້ງການລະລາຍ.ຈະເປັນແນວໃດຖ້າ primers ທີ່ຖືກອອກແບບບໍ່ສາມາດຫຼີກເວັ້ນ primer-dimer?
ວິທີທີ 3: ຫຼຸດຜ່ອນປະລິມານ primers ແລະເພີ່ມອຸນຫະພູມ annealing (ບໍ່ຈໍາເປັນຕ້ອງອະທິບາຍ).
ຄຸນຄ່າທາງປະສາດຂອງອຸນຫະພູມການຫມູນວຽນແມ່ນ 60 ອົງສາ C.ຖ້າຫາກວ່າທ່ານບໍ່ແນ່ໃຈວ່າ, ວິທີການຄັດເລືອກເອົາອຸນຫະພູມ annealing ທີ່ເຫມາະສົມກວ່າ?ຄໍາຕອບແມ່ນຄືກັນກັບການເລືອກຄວາມເຂັ້ມຂົ້ນຂອງ primer -ການທົດສອບ gradient.ເອົາຮູບຈາກບໍລິສັດ Bio-rad ເພື່ອສະແດງໃຫ້ເຫັນບັນຫາ.ສໍາລັບການຂະຫຍາຍຂອງຊິ້ນສ່ວນເປົ້າຫມາຍທີ່ແນ່ນອນ, ກໍານົດແປດ gradients ອຸນຫະພູມ, ແຕ່ລະຄົນມີສາມຊ້ໍາກັນ, ແລະເສັ້ນໂຄ້ງການຂະຫຍາຍທີ່ໄດ້ຮັບແມ່ນດັ່ງຕໍ່ໄປນີ້:
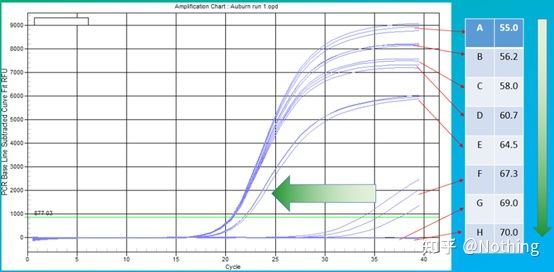
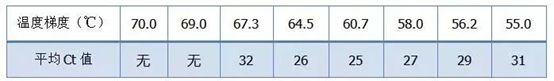
ການຄັດເລືອກອຸນຫະພູມ annealing:
· 70°C, 69°C—ໂດຍພື້ນຖານແລ້ວ, primers ບໍ່ສາມາດປະສົມກັນໄດ້, ສະນັ້ນບໍ່ມີການຂະຫຍາຍ.
· 67.3°C – ມີຈໍານວນຂະຫນາດນ້ອຍຂອງການຂະຫຍາຍຕົວໃນຕອນຕົ້ນ, ແລະຄ່າ Ct ແມ່ນຂ້ອນຂ້າງຫຼາຍ.
·64.5°C—ຄ່າ Ct ຫຼຸດລົງ.
·ຢູ່ທີ່ 60.7 ° C, 58.0 ° C, 56.2 ° C, ແລະ 55.0 ° C, ໂດຍພື້ນຖານແລ້ວຄ່າ Ct ມັກຈະມີຄວາມຫມັ້ນຄົງ, ແຕ່ຄ່າ fluorescence ສຸດທ້າຍແມ່ນແຕກຕ່າງກັນ.
ວິທີການເລືອກ?ຫຼັກການ: ຫຼັກການທໍາອິດແມ່ນຄ່າ Ct ສູງກວ່າ.ສໍາລັບຄ່າ Ct ດຽວກັນ, ເລືອກອຸນຫະພູມ annealing ທີ່ສູງຂຶ້ນເພື່ອຫຼີກເວັ້ນການ dimerization ແລະການຂະຫຍາຍທີ່ບໍ່ສະເພາະ.ເຖິງແມ່ນວ່າມີຄ່າ fluorescence ສູງກວ່າຢູ່ທີ່ 55 ° C, ອາດຈະມີ dimers ຫຼືການຂະຫຍາຍທີ່ບໍ່ສະເພາະຢູ່ໃນມັນ.
ແຕ່ຖ້າທ່ານມີຄວາມສະຫຼາດຄືກັບທ່ານ, ທ່ານແນ່ນອນຈະຄິດວ່າ: ເວົ້າຢ່າງມີເຫດຜົນ, ຖ້າປະຕິກິລິຍາ PCR ແມ່ນສະເພາະຫຼາຍ, ຕາບໃດທີ່ຄວາມເຂັ້ມຂົ້ນຂອງ primer ເກີນຄວາມຕ້ອງການຂັ້ນຕ່ໍາ, ຈຸດສູງແລະຕ່ໍາບໍ່ຄວນມີຜົນກະທົບ, ຄືກັນກັບສີຍ້ອມ fluorescent ແລະ dNTPs.ແທ້ຈິງແລ້ວ, ຕາບໃດທີ່ອຸນຫະພູມ annealing ໄດ້ຖືກປັບປຸງຢ່າງຖືກຕ້ອງ, ຜົນກະທົບຂອງຄວາມເຂັ້ມຂົ້ນຂອງ primer ໃນມູນຄ່າ Ct ຈະຖືກຫຼຸດລົງຕາມທໍາມະຊາດ.
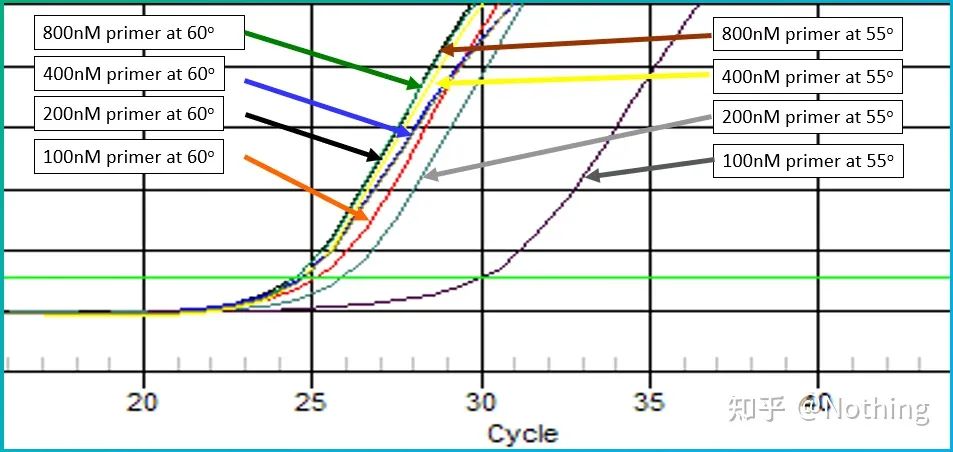
ອຸນຫະພູມ annealing ຖືກປັບໃຫ້ເຫມາະສົມຢ່າງຖືກຕ້ອງ, ແລະຜົນກະທົບຂອງຄວາມເຂັ້ມຂົ້ນ primer ກ່ຽວກັບ CT ຈະຖືກຫຼຸດລົງ
ໂຄງສ້າງຂັ້ນສອງມີຜົນກະທົບຕໍ່ປະສິດທິພາບການຂະຫຍາຍ
ຂໍເອົາຮູບຈາກ Bio-rad ມາສະແດງບັນຫາ.ມັນຍັງອອກແບບ gradient ອຸນຫະພູມເພື່ອຂະຫຍາຍພັນທຸກໍາທີ່ມີໂຄງສ້າງຂັ້ນສອງ.
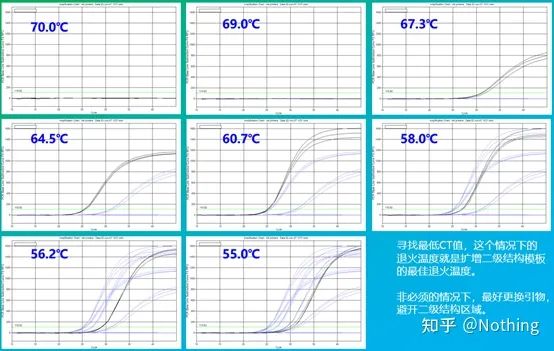
ໂຄງສ້າງຂັ້ນສອງປະກົດຂຶ້ນ
ເຫັນໄດ້ວ່າເມື່ອອຸນຫະພູມຫຼຸດລົງ, ຜະລິດຕະພັນເລີ່ມປະກົດຂຶ້ນ ແລະຄ່າ Ct ກ້າວໄປຂ້າງໜ້າ, ຮອດຄ່າຕໍ່າສຸດຢູ່ທີ່ 60.7 ອົງສາ C, ແລະຫຼັງຈາກນັ້ນເມື່ອອຸນຫະພູມຫຼຸດລົງ, ຄ່າ Ct ຈະໃຫຍ່ຂຶ້ນ.ໃນທາງກົງກັນຂ້າມ, ເມື່ອອຸນຫະພູມເພີ່ມຂຶ້ນ, ໂຄງສ້າງຂັ້ນສອງຈະເປີດຂຶ້ນແລະປະສິດທິພາບການຂະຫຍາຍໃຫຍ່ຂື້ນ.ຫຼັງຈາກເຖິງອຸນຫະພູມທີ່ແນ່ນອນ, ການເພີ່ມອຸນຫະພູມບໍ່ສາມາດປັບປຸງປະສິດທິພາບການຂະຫຍາຍໄດ້.ເນື່ອງຈາກວ່າ primers ບໍ່ສາມາດສົມທົບຢ່າງຫມັ້ນຄົງໃນເວລານີ້.ດັ່ງນັ້ນ,ຊອກຫາອຸນຫະພູມທີ່ມີຄ່າ Ct ຕໍ່າສຸດ, ເຊິ່ງເປັນອຸນຫະພູມທີ່ດີທີ່ສຸດສໍາລັບການຂະຫຍາຍແມ່ແບບໂຄງສ້າງຮອງ!ແນ່ນອນ, ຄົນໂງ່ທີ່ສະຫລາດຕ້ອງຮູ້ວ່າຖ້າມັນບໍ່ຈໍາເປັນ, ມັນດີທີ່ສຸດທີ່ຈະປ່ຽນ primers ແລະຫຼີກເວັ້ນພາກພື້ນໂຄງສ້າງຮອງ.
5. ລະດັບຄໍາຮ້ອງສະຫມັກ
MIQE—ການວິເຄາະຂໍ້ມູນ

ການວິເຄາະຂໍ້ມູນສ່ວນໃຫຍ່ແມ່ນໄດ້ຮັບໂດຍເຄື່ອງມື PCR ປະລິມານ fluorescent.ໃນບົດຄວາມທີ່ຜ່ານມາ, ວຽກງານການວິເຄາະຂໍ້ມູນຈໍານວນຫຼາຍໄດ້ຖືກປະຕິບັດ, ເຊັ່ນ: ການຄວບຄຸມເປົ່າ, ເຊິ່ງໄດ້ຖືກອະທິບາຍໃນການອອກແບບຂອງການທົດລອງ.genes ອ້າງອິງພາຍໃນ, ຕົວເລກຊ້ໍາກັນ, ແລະອື່ນໆ., ໃນທີ່ນີ້ພວກເຮົາສ່ວນໃຫຍ່ອະທິບາຍຄໍາຮ້ອງສະຫມັກຂອງ qPCR.
qPCR ຖືກນໍາໃຊ້ຢ່າງກວ້າງຂວາງ, ແລະການຢັ້ງຢືນການທົດລອງແລະການວິນິດໄສອາຊິດ nucleic ແມ່ນສະຖານະການທີ່ຖືກນໍາໃຊ້ຫຼາຍທີ່ສຸດ.
ປະລິມານຢ່າງແທ້ຈິງ
ບັນທຶກ (ຄວາມເຂັ້ມຂຸ້ນເບື້ອງຕົ້ນ) ມີຄວາມສໍາພັນເສັ້ນກົງກັບຈໍານວນຂອງຮອບວຽນ.ເສັ້ນໂຄ້ງມາດຕະຖານສາມາດຖືກແຕ້ມຈາກມາດຕະຖານທີ່ມີຈໍານວນສໍາເນົາເບື້ອງຕົ້ນທີ່ຮູ້ຈັກ, ນັ້ນແມ່ນ, ການພົວພັນເສັ້ນຂອງຕິກິຣິຍາຂະຫຍາຍສາມາດໄດ້ຮັບ.ອີງຕາມຄ່າ Ct ຂອງຕົວຢ່າງ, ຄວາມເຂັ້ມຂົ້ນໃນຕົວຢ່າງສາມາດຖືກຄິດໄລ່.ຈໍານວນແມ່ແບບທີ່ຈະລວມເອົາ.
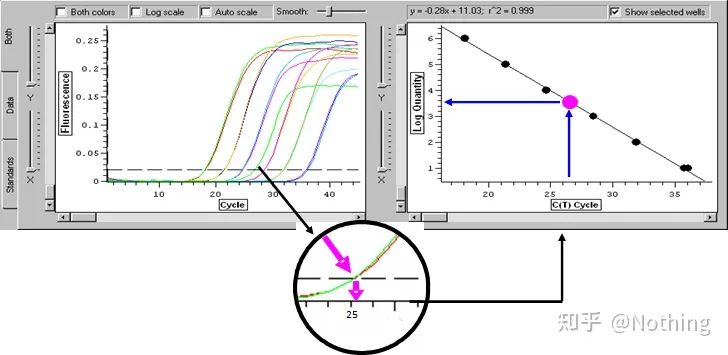
ວິທີການຄິດໄລ່ປະລິມານຢ່າງແທ້ຈິງ
ປະລິມານຢ່າງແທ້ຈິງຕ້ອງອີງໃສ່ເສັ້ນໂຄ້ງມາດຕະຖານ.ເພື່ອເຮັດໃຫ້ເສັ້ນໂຄ້ງມາດຕະຖານ, ມາດຕະຖານແມ່ນຕ້ອງການ.ປົກກະຕິແລ້ວ, ມາດຕະຖານແມ່ນ plasmid ທີ່ໄດ້ຮັບໂດຍການ cloning gene ເປົ້າຫມາຍ.ເປັນຫຍັງມັນເປັນ plasmid?ເນື່ອງຈາກວ່າ plasmid DNA ເປັນວົງກົມແມ່ນມີຄວາມຫມັ້ນຄົງທີ່ສຸດ.ເຈືອຈາງຜະລິດຕະພັນມາດຕະຖານໃນ 5 ຫາ 6 gradients ຕາມອັດຕາສ່ວນສອງເທົ່າ (ການເຈືອຈາງ 10 ເທົ່າ), ແລະເອົາໃຈໃສ່ກັບຄວາມເປັນເອກະພາບໃນເວລາທີ່ diluting.ໃຫ້ຄ່າ Ct ຢູ່ລະຫວ່າງ 15-30.
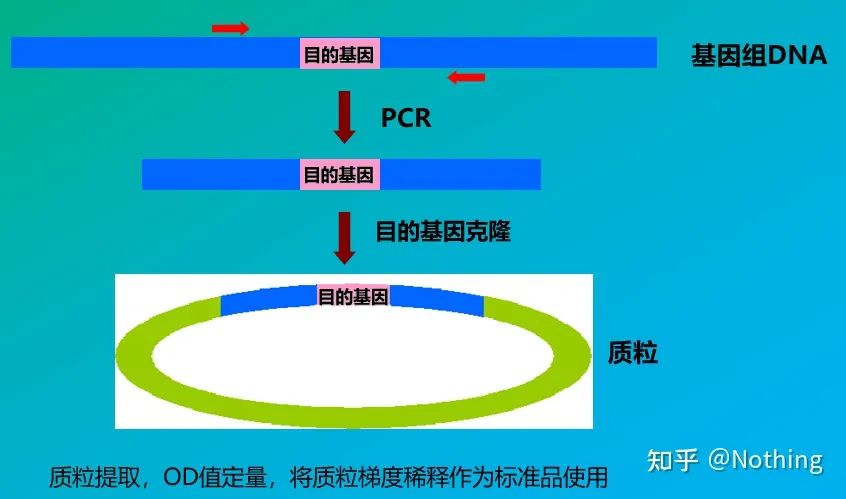
ການກະກຽມມາດຕະຖານ
ໃນຂະນະດຽວກັນ, ຕົວຢ່າງທີ່ຈະທົດສອບຄວນໄດ້ຮັບການເຈືອຈາງຕາມຄວາມເຫມາະສົມ (ຈື່ຈໍາປັດໄຈການເຈືອຈາງ), ແລະຄ່າ Ct ຄວນຫຼຸດລົງລະຫວ່າງ 15-30.ຜະລິດຕະພັນມາດຕະຖານ + ຕົວຢ່າງທີ່ຈະທົດສອບແມ່ນໃສ່ກັບເຄື່ອງຈັກຮ່ວມກັນ.ຫຼັງຈາກແລ່ນ, ເສັ້ນໂຄ້ງມາດຕະຖານໄດ້ຖືກເຮັດດ້ວຍສານມາດຕະຖານ, ແລະຕົວຢ່າງທີ່ຈະທົດສອບໄດ້ຖືກນໍາເຂົ້າໄປໃນເສັ້ນໂຄ້ງມາດຕະຖານເພື່ອຄິດໄລ່ຄວາມເຂັ້ມຂົ້ນ.
ການກວດຫາເຊື້ອໄວຣັສຕັບອັກເສບ B ເຊື້ອໄວຣັສ HBV ເປັນປະລິມານຢ່າງແທ້ຈິງທົ່ວໄປ, ເຊິ່ງສາມາດຄິດໄລ່ຈໍານວນການຄັດລອກເຊື້ອໄວຣັສໃນ 1ml ຂອງເລືອດ.
ການຄິດໄລ່ຈໍານວນສໍາເນົາ
ຄວາມເຂັ້ມຂຸ້ນຂອງຕົວຢ່າງທີ່ຈະທົດສອບ (ng/ul) = OD260 × 50ug/ml × ປັດໄຈການເຈືອຈາງ
ນ້ຳໜັກໂມເລກຸນຕົວຢ່າງ = ຈຳນວນຖານ × 324
ຈໍານວນສໍາເນົາຂອງຕົວຢ່າງທີ່ຈະທົດສອບ (copies/ul) = ຄວາມເຂັ້ມຂຸ້ນຂອງຕົວຢ່າງທີ່ຈະທົດສອບ / ນ້ໍາຫນັກໂມເລກຸນຂອງຕົວຢ່າງ × 6 × 1014

ວິທີການຄິດໄລ່ຕົວເລກສໍາເນົາ
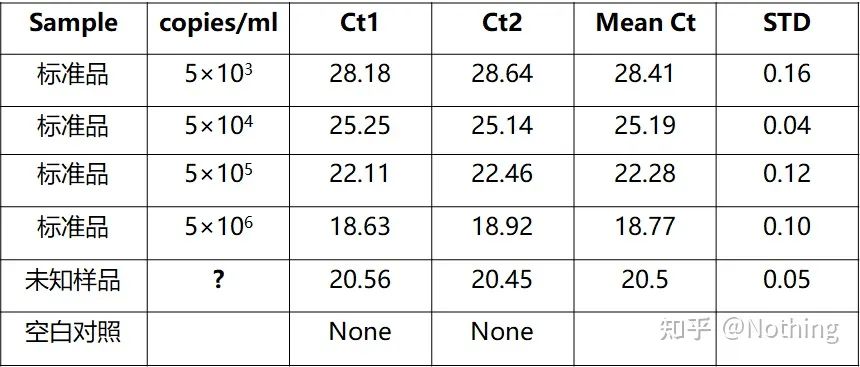
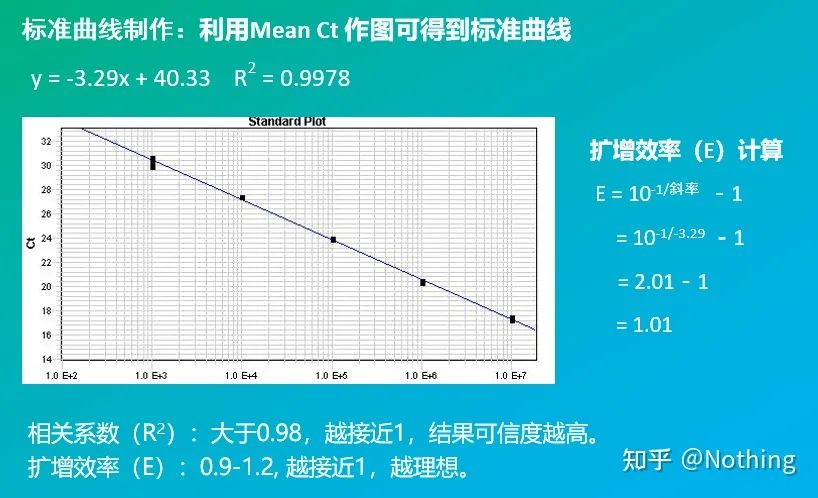

ຂ້າງເທິງນີ້ແມ່ນວິທີການຄິດໄລ່ສໍາລັບການກໍານົດປະລິມານ.ນີ້ແມ່ນບັນຫາທາງຄະນິດສາດທີ່ສາມາດແກ້ໄຂໄດ້ຫຼັງຈາກຮຽນຈົບມັດທະຍົມຕອນຕົ້ນ, ແລະບັນຫາທາງຄະນິດສາດໂດຍທົ່ວໄປແມ່ນແກ້ໄຂດ້ວຍຄອມພິວເຕີ.ຖ້າບໍ່ເຂົ້າໃຈ ສາມາດເຂົ້າມາຕິດຕໍ່ໄດ້.
ປະລິມານທີ່ກ່ຽວຂ້ອງ
ປະລິມານທີ່ກ່ຽວຂ້ອງສ່ວນຫຼາຍແມ່ນໃຊ້ໃນການຄົ້ນຄວ້າວິທະຍາສາດ.ມີເຊື້ອໄວຣັສຫຼາຍປານໃດໃນເລືອດ 1ml, ແລະມັນແມ່ນເຊື້ອໄວຣັສ DNA, ນີ້ແມ່ນເຫດການທີ່ຂ້ອນຂ້າງກໍານົດ: ຈໍານວນເລືອດສາມາດກໍານົດໄດ້, ແລະເຊື້ອໄວຣັສ DNA ແມ່ນຂ້ອນຂ້າງຄົງທີ່.ຢ່າງໃດກໍ່ຕາມ, ມັນເປັນການຍາກສໍາລັບພວກເຮົາທີ່ຈະປຽບທຽບຈໍານວນການຖອດຂໍ້ຄວາມຂອງພັນທຸກໍາໃນໃບ, ເພາະວ່າມັນຍາກທີ່ຈະກໍານົດຂະຫນາດ, ນ້ໍາຫນັກ, ແລະຄວາມອ່ອນໂຍນຂອງໃບ, ຈໍານວນ RNA ທີ່ສະກັດອອກແມ່ນຍາກທີ່ຈະກໍານົດ, ແລະປະສິດທິພາບຂອງ reverse transcription ຍັງຍາກທີ່ຈະກໍານົດ, ນັ້ນແມ່ນ, ຂັ້ນຕອນໃດກໍ່ຕາມອາດຈະເຮັດໃຫ້ຂໍ້ມູນທົດລອງມີຂໍ້ບົກພ່ອງແລະບໍ່ສາມາດນໍາໃຊ້ໄດ້.
ດັ່ງນັ້ນ, ປະລິມານທີ່ກ່ຽວຂ້ອງຕ້ອງແນະນໍາອົງປະກອບ:gene ອ້າງອີງພາຍໃນ.
ໃນຄໍາສັບຕ່າງໆອື່ນໆ, ປະລິມານທີ່ກ່ຽວຂ້ອງແມ່ນຕົວຈິງແລ້ວເປັນການປຽບທຽບລະຫວ່າງ gene ເປົ້າຫມາຍແລະ gene ອ້າງອີງພາຍໃນ.ເມື່ອປຽບທຽບຢູ່ໃນເນື້ອເຍື່ອດຽວກັນແລະຈຸລັງດຽວກັນ, ອິດທິພົນຂອງຂະຫນາດຕົວຢ່າງ, ຈໍານວນການສະກັດເອົາ RNA, ປະສິດທິພາບການຖອດຂໍ້ຄວາມແບບປີ້ນກັບກັນ, ແລະປະສິດທິພາບ PCR ແມ່ນຂ້ອນຂ້າງນ້ອຍ.ເນື່ອງຈາກຂະຫນາດຕົວຢ່າງຂະຫນາດນ້ອຍ, ທັງພັນທຸກໍາອ້າງອີງພາຍໃນແລະ genes ເປົ້າຫມາຍໄດ້ຖືກຫຼຸດລົງຂ້ອນຂ້າງ.ຍ້ອນເຫດນີ້ພວກເຮົາໄດ້ເນັ້ນໜັກເຖິງຄວາມເປັນເອກະພາບ ແລະ ສະຖຽນລະພາບກ່ອນນີ້.
genes ອ້າງອີງພາຍໃນໂດຍທົ່ວໄປແມ່ນພັນທຸ ກຳ ຮັກສາເຮືອນ(house-keeping genes), ເຊິ່ງຫມາຍເຖິງປະເພດຂອງ genes ທີ່ສະແດງອອກຢ່າງຫມັ້ນຄົງໃນທຸກຈຸລັງ, ແລະຜະລິດຕະພັນຂອງມັນເປັນສິ່ງຈໍາເປັນເພື່ອຮັກສາກິດຈະກໍາຊີວິດພື້ນຖານຂອງຈຸລັງ.
ຢ່າສັບສົນແນວຄວາມຄິດນີ້.genes ຮັກສາເຮືອນແມ່ນຂໍ້ກໍານົດການເຮັດວຽກທາງຊີວະພາບ, ໃນຂະນະທີ່ genes ອ້າງອິງພາຍໃນແມ່ນຂໍ້ກໍານົດດ້ານວິຊາການທົດລອງ.ພັນທຸ ກຳ ການຮັກສາເຮືອນ ຈຳ ເປັນຕ້ອງຜ່ານການກວດສອບກ່ອນທີ່ພວກມັນຈະຖືກເລືອກເປັນພັນທຸ ກຳ ອ້າງອີງພາຍໃນ.
ຕົວຢ່າງເຊັ່ນ, ພວກເຮົາໄດ້ເລືອກພັນທຸກໍາໃນຄົວເຮືອນໃນຮູບຂ້າງລຸ່ມນີ້ເພື່ອທົດສອບລະດັບການສະແດງອອກຂອງພວກເຂົາໃນຈຸລັງເນື້ອເຍື່ອທີ່ແຕກຕ່າງກັນ, ແລະພົບວ່າລະດັບການສະແດງອອກຂອງ β-2-microglobulin ແມ່ນຂ້ອນຂ້າງແຕກຕ່າງຈາກ genes ຂອງສາມອື່ນໆ, ດັ່ງນັ້ນພວກມັນບໍ່ສາມາດຖືກນໍາໃຊ້ເປັນ genes ອ້າງອີງພາຍໃນ.
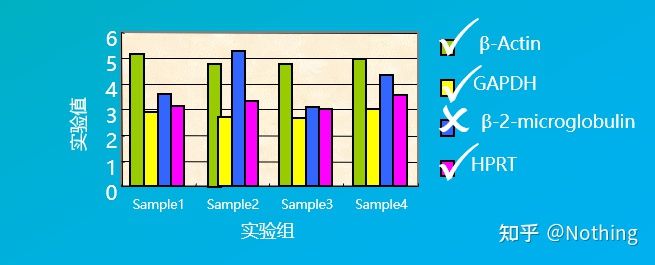
ຫຼັງຈາກເຂົ້າໃຈການທໍາງານການແກ້ໄຂຂອງ gene ອ້າງອິງພາຍໃນ, ສອງ algorithms ແມ່ນມາຈາກການແນະນໍາຂອງ gene ອ້າງອິງພາຍໃນ.
·ວິທີການເສັ້ນໂຄ້ງມາດຕະຖານສອງເທົ່າ
·2 – △△ Ct method (ວິທີປຽບທຽບຄ່າ CT)
ຖ້າຫາກວ່າທ່ານມີຄວາມສົນໃຈໃນການສຶກສາຊະນິດພັນແລະຫນ້າທີ່ຂອງ gene, ກະລຸນາຍົກເລີກການຄົ້ນຄວ້າກ່ຽວກັບ algorithms ແລະນໍາໃຊ້ສູດໂດຍກົງ, ຫຼືນໍາໃຊ້ເຄື່ອງຈັກໂດຍກົງ;ຖ້າທ່ານເປັນຄົນຊື່ກົງໃນຄະນິດສາດແລະວິສະວະກໍາ, ກະລຸນາຮູ້ສຶກວ່າບໍ່ເສຍຄ່າ.
ວິທີການເສັ້ນໂຄ້ງມາດຕະຖານສອງເທົ່າ
ຄິດໄລ່ gene ເປົ້າຫມາຍແລະ gene ຮັກສາເຮືອນຂອງຕົວຢ່າງການຄວບຄຸມແລະຕົວຢ່າງທີ່ຈະທົດສອບໂດຍຜ່ານເສັ້ນໂຄ້ງມາດຕະຖານ, ແລະຫຼັງຈາກນັ້ນຄິດໄລ່ມູນຄ່າພີ່ນ້ອງຕາມສູດການຄິດໄລ່, ຊຶ່ງເປັນລະດັບການສະແດງອອກຂອງພີ່ນ້ອງ.
ຂໍ້ໄດ້ປຽບ: ການວິເຄາະງ່າຍດາຍ, ການເພີ່ມປະສິດທິພາບການທົດລອງຂ້ອນຂ້າງງ່າຍດາຍ
ຂໍ້ເສຍ: ສໍາລັບແຕ່ລະ gene, ແຕ່ລະຮອບຂອງການທົດລອງຈະຕ້ອງເຮັດໃຫ້ເສັ້ນໂຄ້ງມາດຕະຖານ
ການນໍາໃຊ້: ຫນຶ່ງໃນສອງວິທີການປະລິມານທີ່ຖືກຮັບຮູ້ຫຼາຍທີ່ສຸດໃນການສຶກສາລະບຽບການສະແດງອອກຂອງເຊື້ອສາຍ
ສູດດັ່ງຕໍ່ໄປນີ້:

ຕົວຢ່າງມີດັ່ງນີ້:
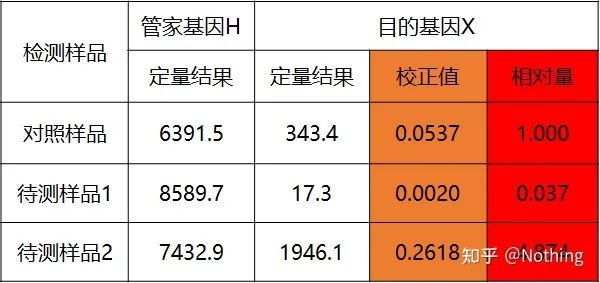
ຄິດໄລ່ຈໍານວນທີ່ສົມທຽບໂດຍອີງໃສ່ຜົນໄດ້ຮັບປະລິມານ
2 – △△ Ct method (ວິທີປຽບທຽບຄ່າ CT)

ຂໍ້ດີ: ບໍ່ຈໍາເປັນຕ້ອງເຮັດເສັ້ນໂຄ້ງມາດຕະຖານ
ຂໍ້ເສຍ: ມັນສົມມຸດວ່າປະສິດທິພາບການຂະຫຍາຍແມ່ນຢູ່ໃກ້ກັບ 100%;ມາດຕະຖານ deviation ແມ່ນ <5%, ແລະເສັ້ນໂຄ້ງມາດຕະຖານແລະປະສິດທິພາບລະຫວ່າງການຂະຫຍາຍແຕ່ລະແມ່ນສົມມຸດຕິຖານ;ການເພີ່ມປະສິດທິພາບຂອງເງື່ອນໄຂການທົດລອງແມ່ນສັບສົນຫຼາຍ.
ການນໍາໃຊ້: ຫນຶ່ງໃນສອງວິທີການປະລິມານທີ່ຖືກຮັບຮູ້ຫຼາຍທີ່ສຸດໃນການສຶກສາລະບຽບການສະແດງອອກຂອງເຊື້ອສາຍ
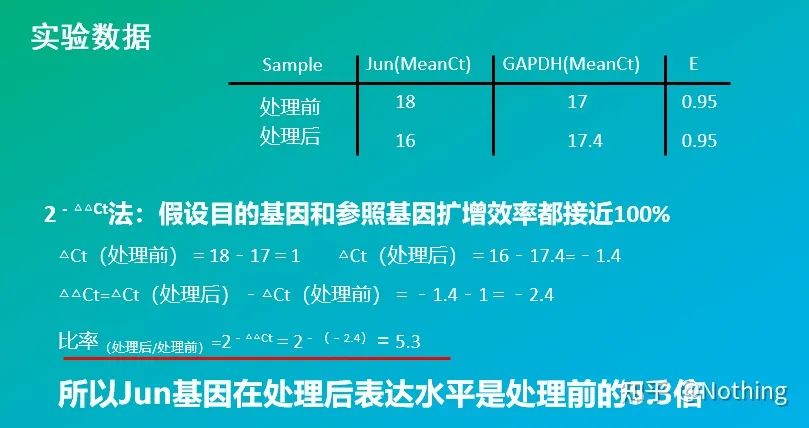
ແນ່ນອນ, ປະສິດທິພາບການຂະຫຍາຍແມ່ນປົກກະຕິແລ້ວເປັນໄປບໍ່ໄດ້ທີ່ຈະສົມບູນແບບ 1. ວິທີການແກ້ໄຂ: ຖ້າພວກເຮົາຮູ້ວ່າ gene ເປົ້າຫມາຍແລະ gene ອ້າງອິງມີປະສິດທິພາບການຂະຫຍາຍຄືກັນ, ແຕ່ປະສິດທິພາບການຂະຫຍາຍບໍ່ເທົ່າກັບ 1, ຫຼັງຈາກນັ້ນ 2-△△Ct ສາມາດແກ້ໄຂໄດ້ຄື: (1+E)-△△ Ct. ຕົວຢ່າງ 0. ປະສິດທິພາບແມ່ນ 9, calculation. ເຖິງ 1.95-△△ Ct
ມາຮອດປະຈຸ, ເນື້ອໃນກ່ຽວກັບ PCR ປະລິມານ fluorescent ໄດ້ສິ້ນສຸດລົງ.
ເວລາປະກາດ: ເມສາ-06-2023



