



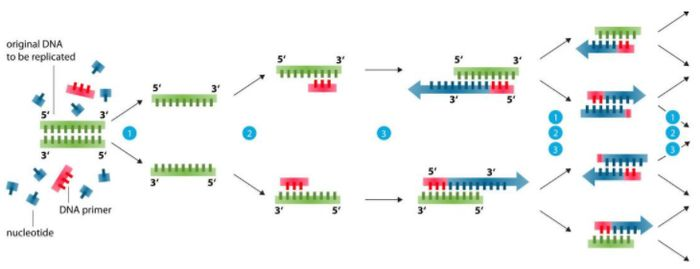
ພື້ນຖານການອອກແບບ primer (99% ບັນຫາສາມາດແກ້ໄຂໄດ້)
1. ຄວາມຍາວຂອງ primer: ປື້ມແບບຮຽນຕ້ອງການ 15-30bp, ປົກກະຕິແລ້ວປະມານ 20bp.ສະພາບຕົວຈິງແມ່ນດີກວ່າທີ່ຈະເປັນ 18-24bp ເພື່ອຮັບປະກັນສະເພາະ, ແຕ່ຕໍ່ໄປອີກແລ້ວທີ່ດີກວ່າ, primer ຍາວເກີນໄປຍັງຈະຫຼຸດຜ່ອນຄວາມສະເພາະ, ແລະຫຼຸດຜ່ອນຜົນຜະລິດ.
2. Primer amplification span: 200-500bp ແມ່ນເຫມາະສົມ, ແລະ fragment ສາມາດຂະຫຍາຍໄດ້ເຖິງ 10kb ພາຍໃຕ້ເງື່ອນໄຂສະເພາະ.
3. ພື້ນຖານ primer: ເນື້ອໃນຂອງ G + C ຄວນຈະເປັນ 40-60%, ຜົນກະທົບການຂະຫຍາຍ G + C ຫນ້ອຍເກີນໄປບໍ່ດີ, G + C ຫຼາຍເກີນໄປແມ່ນງ່າຍທີ່ຈະປາກົດແຖບທີ່ບໍ່ແມ່ນສະເພາະ.ATGC ແມ່ນແຈກຢາຍແບບສຸ່ມທີ່ດີທີ່ສຸດ, ຫຼີກເວັ້ນການເປັນກຸ່ມຂອງຫຼາຍກວ່າ 5 purine ຫຼື pyrimidine nucleotides.Multi-gc ສໍາລັບລໍາດັບ 5′ ແລະລະດັບກາງເພື່ອເພີ່ມຄວາມຫມັ້ນຄົງ, ຫຼີກເວັ້ນການອຸດົມສົມບູນ GC ໃນຕອນທ້າຍຂອງ 3′, ບໍ່ມີ GC ສໍາລັບຖານ 3 ສຸດທ້າຍ, ຫຼືບໍ່ມີ GC ສໍາລັບ 3 ຂອງ 5 ພື້ນຖານສຸດທ້າຍ.
4. ຫຼີກເວັ້ນການໂຄງສ້າງຂັ້ນສອງໃນ primers, ແລະຫຼີກເວັ້ນການປະສົມລະຫວ່າງສອງ primers, ໂດຍສະເພາະແມ່ນການເສີມຢູ່ປາຍ 3 ', ຖ້າບໍ່ດັ່ງນັ້ນ primer dimer ຈະຖືກສ້າງຕັ້ງຂຶ້ນແລະແຖບ amplified ທີ່ບໍ່ແມ່ນສະເພາະຈະຖືກສ້າງຂຶ້ນ.
5. ຖານທີ່ 3 'ໃນຕອນທ້າຍຂອງ primers, ໂດຍສະເພາະແມ່ນຖານສຸດທ້າຍແລະ penultimate, ຄວນໄດ້ຮັບການຈັບຄູ່ຢ່າງເຂັ້ມງວດເພື່ອຫຼີກເວັ້ນການ PCR ຄວາມລົ້ມເຫຼວເນື່ອງຈາກພື້ນຖານຂອງ terminal unpaired.
6. primers ມີຫຼືສາມາດເພີ່ມດ້ວຍສະຖານທີ່ cleavage ທີ່ເຫມາະສົມ, ແລະລໍາດັບເປົ້າຫມາຍທີ່ຂະຫຍາຍໄດ້ຄວນຈະມີສະຖານທີ່ cleavage ທີ່ເຫມາະສົມ, ເຊິ່ງເປັນປະໂຫຍດຫຼາຍສໍາລັບການວິເຄາະ cleavage ຫຼື molecular cloning.
7. ຄວາມສະເພາະຂອງ primers: primers ຄວນຈະບໍ່ມີ homology ຊັດເຈນກັບລໍາດັບອື່ນໆໃນຖານຂໍ້ມູນລໍາດັບອາຊິດ nucleic.
8. ຮຽນຮູ້ການນໍາໃຊ້ຊອບແວ: PP5, Oligo6, DNAstar, Vector NTI, primer3 (ການອອກແບບອອນໄລນ໌ນີ້ເຮັດວຽກທີ່ດີທີ່ສຸດ).
ເນື້ອໃນຂ້າງເທິງສາມາດແກ້ໄຂຢ່າງຫນ້ອຍ 99% ຂອງບັນຫາການອອກແບບ primer.
ຄວບຄຸມລາຍລະອຽດຂອງການອອກແບບ primer
1. ຄວາມຍາວຂອງ primer
ຄວາມຍາວ primer ທົ່ວໄປແມ່ນ 18-30 ຖານ.ໂດຍທົ່ວໄປ, ປັດໃຈທີ່ສໍາຄັນທີ່ສຸດໃນການກໍານົດອຸນຫະພູມການຫມູນວຽນຂອງ primer ແມ່ນຄວາມຍາວຂອງ primer.ອຸນຫະພູມ annealing ຂອງ primer ໂດຍທົ່ວໄປແມ່ນເລືອກ (ຄ່າ Tm -5 ℃), ແລະບາງການນໍາໃຊ້ໂດຍກົງຄ່າ Tm.ສູດຕໍ່ໄປນີ້ສາມາດຖືກໃຊ້ເພື່ອຄິດໄລ່ປະມານອຸນຫະພູມການຫມູນວຽນຂອງ primers.
ເມື່ອຄວາມຍາວຂອງ primer ຫນ້ອຍກວ່າ 20bp: [4(G+C)+2(A+T)]-5℃
ເມື່ອຄວາມຍາວຂອງ primer ຫຼາຍກວ່າ 20bp: 62.3℃+0.41℃(%GC)-500/length-5℃
ນອກຈາກນັ້ນ, ຊອບແວຈໍານວນຫຼາຍຍັງສາມາດຖືກນໍາໃຊ້ເພື່ອຄິດໄລ່ອຸນຫະພູມ annealing, ຫຼັກການການຄິດໄລ່ຈະແຕກຕ່າງກັນ, ດັ່ງນັ້ນບາງຄັ້ງມູນຄ່າການຄິດໄລ່ອາດຈະມີຊ່ອງຫວ່າງເລັກນ້ອຍ.ເພື່ອເພີ່ມປະສິດທິພາບປະຕິກິລິຍາ PCR, primers ສັ້ນທີ່ສຸດທີ່ຮັບປະກັນອຸນຫະພູມບໍ່ຫນ້ອຍກວ່າ 54 ℃ແມ່ນຖືກນໍາໃຊ້ເພື່ອປະສິດທິພາບແລະສະເພາະທີ່ດີທີ່ສຸດ.
ໂດຍລວມແລ້ວ, ຄວາມສະເພາະຂອງ primer ເພີ່ມຂຶ້ນໂດຍປັດໄຈສີ່ສໍາລັບແຕ່ລະ nucleotide ເພີ່ມເຕີມ, ດັ່ງນັ້ນຄວາມຍາວ primer ຕໍາ່ສຸດທີ່ສໍາລັບຄໍາຮ້ອງສະຫມັກສ່ວນໃຫຍ່ແມ່ນ 18 nucleotides.ຂອບເຂດຈໍາກັດເທິງຂອງຄວາມຍາວ primer ແມ່ນບໍ່ສໍາຄັນຫຼາຍ, ສ່ວນໃຫຍ່ແມ່ນກ່ຽວຂ້ອງກັບປະສິດທິພາບຕິກິຣິຍາ.ເນື່ອງຈາກວ່າ entropy, primer ຍາວກວ່າ, ອັດຕາທີ່ຕໍ່າກວ່າທີ່ມັນຍຶດຫມັ້ນກັບ DNA ເປົ້າຫມາຍເພື່ອສ້າງເປັນແມ່ແບບສອງສາຍທີ່ຫມັ້ນຄົງສໍາລັບ DNA polymerase ທີ່ຈະຜູກມັດ.
ເມື່ອນໍາໃຊ້ຊອບແວໃນການອອກແບບ primers, ຄວາມຍາວຂອງ primers ສາມາດຖືກກໍານົດໂດຍມູນຄ່າ TM, ໂດຍສະເພາະສໍາລັບ primers fluorescence quantitative PCR, TM = 60 ℃ຫຼືດັ່ງນັ້ນຄວນຈະຖືກຄວບຄຸມ.
2.GC ເນື້ອໃນ
ໂດຍທົ່ວໄປ, ເນື້ອໃນຂອງ G + C ໃນລໍາດັບ primer ແມ່ນ 40% ~ 60%, ແລະເນື້ອໃນ GC ແລະມູນຄ່າ Tm ຂອງຄູ່ primers ຄວນຖືກປະສານງານ.ຖ້າ primer ມີແນວໂນ້ມ GC ຫຼື AT ທີ່ຮ້າຍແຮງ, ຈໍານວນທີ່ເຫມາະສົມຂອງ A, T ຫຼື G ແລະ C ຫາງສາມາດຖືກເພີ່ມໃສ່ 5 'ໃນຕອນທ້າຍຂອງ primer.
3. ອຸນຫະພູມ Annealing
ອຸນຫະພູມ annealing ຄວນ 5 ℃ຕ່ໍາກ່ວາອຸນຫະພູມ unchain.ຖ້າຈໍານວນຂອງພື້ນຖານ primer ມີຂະຫນາດນ້ອຍ, ອຸນຫະພູມ annealing ສາມາດເພີ່ມຂຶ້ນຢ່າງເຫມາະສົມ, ເຊິ່ງສາມາດເພີ່ມຄວາມສະເພາະຂອງ PCR.ຖ້າຈໍານວນຂອງຖານມີຂະຫນາດໃຫຍ່, ອຸນຫະພູມ annealing ສາມາດຫຼຸດລົງໄດ້ຢ່າງເຫມາະສົມ.ຄວາມແຕກຕ່າງຂອງອຸນຫະພູມ annealing ລະຫວ່າງຄູ່ຂອງ primers ຂອງ 4℃ ~ 6℃ ຈະບໍ່ມີຜົນກະທົບຜົນຜະລິດ PCR, ແຕ່ໂດຍສະເພາະອຸນຫະພູມ annealing ຂອງ primers ຄູ່ແມ່ນຄືກັນ, ຊຶ່ງສາມາດແຕກຕ່າງກັນລະຫວ່າງ 55 ℃ ~ 75 ℃.
4. ຫຼີກເວັ້ນພື້ນທີ່ໂຄງສ້າງຮອງຂອງແມ່ແບບການຂະຫຍາຍ
ມັນດີທີ່ສຸດທີ່ຈະຫຼີກເວັ້ນພາກພື້ນໂຄງສ້າງຮອງຂອງແມ່ແບບໃນເວລາທີ່ເລືອກຊິ້ນສ່ວນຂະຫຍາຍ.ໂຄງສ້າງຂັ້ນສອງທີ່ຫມັ້ນຄົງຂອງຊິ້ນສ່ວນເປົ້າຫມາຍສາມາດຄາດຄະເນແລະຄາດຄະເນໂດຍຊອບແວຄອມພິວເຕີທີ່ກ່ຽວຂ້ອງ, ເຊິ່ງເປັນປະໂຫຍດສໍາລັບການເລືອກແມ່ແບບ.ຜົນໄດ້ຮັບຈາກການທົດລອງສະແດງໃຫ້ເຫັນວ່າການຂະຫຍາຍຕົວມັກຈະບໍ່ປະສົບຜົນສໍາເລັດເມື່ອພະລັງງານຟຣີ (△G) ຂອງພາກພື້ນທີ່ຈະຂະຫຍາຍແມ່ນຫນ້ອຍກວ່າ 58.6lkJ/mol.
5. ບໍ່ກົງກັນກັບ DNA ເປົ້າໝາຍ
ເມື່ອ ລຳ ດັບ DNA ເປົ້າ ໝາຍ ຂະຫຍາຍໃຫຍ່ຂື້ນ, primer ອາດຈະຜູກມັດກັບຫຼາຍພາກສ່ວນຂອງ DNA ເປົ້າ ໝາຍ, ເຮັດໃຫ້ມີແຖບຫຼາຍປະກົດຢູ່ໃນຜົນໄດ້ຮັບ.ເວລານີ້ມັນຈໍາເປັນຕ້ອງໃຊ້ການທົດສອບຊອບແວ BLAST, ເວັບໄຊທ໌:http://www.ncbi.nlm.nih.gov/BLAST/.ເລືອກການຈັດຮຽງສອງລໍາດັບ (bl2seq).
ການວາງລໍາດັບ primer ໄປຫາເຂດ 1 ແລະເປົ້າຫມາຍລໍາດັບ DNA ໄປຫາເຂດ 2 ແມ່ນສາມາດແລກປ່ຽນກັນໄດ້, ແລະ BLAST ຄິດໄລ່ຄວາມສົມດຸນ, antisense, ແລະຄວາມເປັນໄປໄດ້ອື່ນໆ, ດັ່ງນັ້ນຜູ້ໃຊ້ບໍ່ຈໍາເປັນຕ້ອງສັງເກດເຫັນວ່າທັງສອງຕ່ອງໂສ້ແມ່ນຕ່ອງໂສ້ຄວາມຮູ້ສຶກ.ທ່ານຍັງສາມາດໃສ່ຕົວເລກ GI ຖ້າທ່ານຮູ້ຈໍານວນ GI ຂອງລໍາດັບໃນຖານຂໍ້ມູນ, ດັ່ງນັ້ນທ່ານບໍ່ຈໍາເປັນຕ້ອງວາງສ່ວນໃຫຍ່ຂອງລໍາດັບ.ສຸດທ້າຍ, ຄລິກຈັດຮຽງຢູ່ 3 ເພື່ອເບິ່ງວ່າ primer ມີຫຼາຍສະຖານທີ່ homologous ໃນ DNA ເປົ້າຫມາຍ.
6. Primer terminal
ປາຍ 3 'ຂອງ primer ແມ່ນບ່ອນທີ່ການຂະຫຍາຍເລີ່ມຕົ້ນ, ສະນັ້ນມັນເປັນສິ່ງສໍາຄັນທີ່ຈະປ້ອງກັນບໍ່ໃຫ້ເກີດຄວາມຜິດປົກກະຕິຈາກການເລີ່ມຕົ້ນ.ປາຍ 3 'ບໍ່ຄວນມີຫຼາຍກວ່າ 3 G ຫຼື C ຕິດຕໍ່ກັນ, ເພາະວ່ານີ້ຈະເຮັດໃຫ້ primer ຖືກກະຕຸ້ນຜິດພາດໃນພາກພື້ນລໍາດັບ G+C enrichment.ປາຍ 3 'ບໍ່ສາມາດສ້າງໂຄງສ້າງຮອງໃດໆ, ຍົກເວັ້ນໃນປະຕິກິລິຍາ PCR ພິເສດ (AS-PCR), 3' ປາຍຂອງ primer ບໍ່ສາມາດບໍ່ກົງກັນໄດ້.ຕົວຢ່າງ, ຖ້າເຂດການເຂົ້າລະຫັດຖືກຂະຫຍາຍ, 3 'ສິ້ນສຸດຂອງ primer ບໍ່ຄວນຖືກຢຸດຢູ່ທີ່ຕໍາແຫນ່ງທີສາມຂອງ codon, ເພາະວ່າຕໍາແຫນ່ງທີ່ສາມຂອງ codon ມີຄວາມສ່ຽງທີ່ຈະ degenerate, ເຊິ່ງຈະມີຜົນກະທົບສະເພາະແລະປະສິດທິພາບຂອງການຂະຫຍາຍ.ເມື່ອໃຊ້ primers ຊ້ອນກັນ, ອ້າງອີງໃສ່ຕາຕະລາງການນໍາໃຊ້ codon, ເອົາໃຈໃສ່ກັບຄວາມມັກທາງຊີວະພາບ, ຫ້າມໃຊ້ primers ຊ້ອນທ້າຍ 3′, ແລະໃຊ້ primers ທີ່ມີຄວາມເຂັ້ມຂົ້ນສູງກວ່າ (1uM-3uM).
7. ໂຄງສ້າງຮອງຂອງ primers
primers ຕົວເອງບໍ່ຄວນມີລໍາດັບເສີມ, ຖ້າບໍ່ດັ່ງນັ້ນ primers ຕົວເອງຈະພັບເຂົ້າໄປໃນໂຄງສ້າງຂອງ hairpin, ແລະໂຄງສ້າງຮອງນີ້ຈະມີຜົນກະທົບຕໍ່ການຜູກມັດຂອງ primers ແລະ templates ເນື່ອງຈາກການຂັດຂວາງ steric.ຖ້າການຕັດສິນປອມຖືກນໍາໃຊ້, ພື້ນຖານການເສີມຢ່າງຕໍ່ເນື່ອງຂອງ primers ຕົວເອງບໍ່ຄວນສູງກວ່າ 3bp.ບໍ່ຄວນມີການປະສົມກັນລະຫວ່າງສອງ primers, ໂດຍສະເພາະແມ່ນການຊ້ອນກັນຂອງ 3 'ທ້າຍຄວນໄດ້ຮັບການຫຼີກເວັ້ນເພື່ອປ້ອງກັນການສ້າງ primer dimers.ໂດຍທົ່ວໄປແລ້ວ, ບໍ່ຄວນມີຫຼາຍກວ່າ 4 ພື້ນຖານທີ່ຕິດກັນ ຫຼື ຄວາມສົມບູນລະຫວ່າງຄູ່ຂອງ primers.
8. ເພີ່ມເຄື່ອງຫມາຍຫຼື loci
The 5 'end ມີຜົນກະທົບເລັກນ້ອຍຕໍ່ສະເພາະການຂະຫຍາຍໃຫຍ່ຂື້ນແລະດັ່ງນັ້ນຈຶ່ງສາມາດດັດແປງໄດ້ໂດຍບໍ່ມີຜົນກະທົບຕໍ່ຄວາມສະເພາະຂອງ amplification.ການດັດແກ້ຂອງ primer 5 'ສິ້ນສຸດປະກອບມີ: ເພີ່ມສະຖານທີ່ຈໍາກັດ enzyme;ປ້າຍຊື່ biotin, fluorescence, digoxin, Eu3+, ແລະອື່ນໆ. ແນະນໍາການຜູກມັດຂອງທາດໂປຼຕີນຈາກລໍາດັບ DNA;ການແນະນໍາສະຖານທີ່ກາຍພັນ, ການແຊກແລະຂາດລໍາດັບການກາຍພັນແລະການແນະນໍາລໍາດັບການສົ່ງເສີມ, ແລະອື່ນໆ, ພື້ນຖານເພີ່ມເຕີມຈະຫຼາຍຫຼືຫນ້ອຍຜົນກະທົບຕໍ່ປະສິດທິພາບຂອງ amplification ແລະເພີ່ມໂອກາດຂອງການສ້າງ primer dimer, ແຕ່ການສໍາປະທານບາງຢ່າງຕ້ອງເຮັດສໍາລັບຂັ້ນຕອນຕໍ່ໄປ.ລໍາດັບເພີ່ມເຕີມທີ່ບໍ່ມີຢູ່ໃນລໍາດັບເປົ້າຫມາຍ, ເຊັ່ນ: ສະຖານທີ່ຈໍາກັດແລະລໍາດັບການໂຄສະນາ, ສາມາດຖືກເພີ່ມໃສ່ 5′ ທ້າຍຂອງ primer ໂດຍບໍ່ມີຜົນກະທົບຕໍ່ສະເພາະ.ລໍາດັບເຫຼົ່ານີ້ບໍ່ໄດ້ຖືກລວມເຂົ້າໃນການຄິດໄລ່ຄ່າ primer Tm, ແຕ່ຄວນໄດ້ຮັບການທົດສອບສໍາລັບຄວາມສົມບູນແລະໂຄງສ້າງຮອງພາຍໃນ.
9. subclones
ສ່ວນຫຼາຍແລ້ວ, PCR ແມ່ນພຽງແຕ່ cloning ເບື້ອງຕົ້ນ, ແລະຫຼັງຈາກນັ້ນພວກເຮົາຈໍາເປັນຕ້ອງ subclone ຊິ້ນສ່ວນເປົ້າຫມາຍເຂົ້າໄປໃນ vectors ຕ່າງໆ, ດັ່ງນັ້ນພວກເຮົາຈໍາເປັນຕ້ອງອອກແບບພື້ນຖານເພີ່ມເຕີມສໍາລັບການປະຕິບັດງານຕໍ່ໄປໃນຂັ້ນຕອນ PCR.
ບາງລໍາດັບທີ່ຖືກອອກແບບມາສໍາລັບການຍ່ອຍຍ່ອຍແມ່ນສະຫຼຸບຂ້າງລຸ່ມນີ້.
ສະຖານທີ່ຈໍາກັດ endonuclease ໄດ້ຖືກເພີ່ມ
ການເພີ່ມສະຖານທີ່ຈໍາກັດ enzyme ແມ່ນວິທີການທີ່ໃຊ້ທົ່ວໄປທີ່ສຸດສໍາລັບການຍ່ອຍຜະລິດຕະພັນ PCR.ໂດຍທົ່ວໄປ, ສະຖານທີ່ cleavage ແມ່ນຫົກຖານ, ນອກເຫນືອໄປຈາກ 5 'ໃນຕອນທ້າຍຂອງສະຖານທີ່ cleavage ຈໍາເປັນຕ້ອງໄດ້ເພີ່ມ 2 ~ 3 ຖານປ້ອງກັນ.ຢ່າງໃດກໍ່ຕາມ, ຈໍານວນຂອງຖານປ້ອງກັນທີ່ຕ້ອງການໂດຍ enzymes ທີ່ແຕກຕ່າງກັນແມ່ນແຕກຕ່າງກັນ.ຕົວຢ່າງ, SalⅠ ຕ້ອງການບໍ່ມີຖານປ້ອງກັນ, EcoRⅤ ຕ້ອງການ 1 ຖານປ້ອງກັນ, NotⅠ ຕ້ອງການ 2 ຖານປ້ອງກັນ, ແລະ Hind Ⅲ ຕ້ອງການ 3 ຖານປ້ອງກັນ.
LIC ເພີ່ມຫາງ
ຊື່ເຕັມຂອງ LIC ແມ່ນ Ligation-Independent cloning, ເປັນວິທີການໂຄນນິນທີ່ປະດິດໂດຍ Navogen ໂດຍສະເພາະສໍາລັບພາກສ່ວນຂອງ vector pET.ຜູ້ໃຫ້ບໍລິການ pET ທີ່ກະກຽມໂດຍວິທີ LIC ມີ 12-15 ພື້ນຖານຫນຽວດຽວປາຍ, ເຊິ່ງເຮັດໃຫ້ປາຍຫນຽວທີ່ສອດຄ້ອງກັນໃສ່ຊິ້ນສ່ວນທີ່ໃສ່ເປົ້າຫມາຍ.ເພື່ອຈຸດປະສົງການຂະຫຍາຍ, ລຳດັບ primer 5′ ຂອງຊິ້ນສ່ວນທີ່ໃສ່ຄວນເສີມ vector LIC.ກິດຈະກໍາພິເສດຂອງ 3′→5′ ຂອງ T4 DNA polymerase ສາມາດປະກອບເປັນເສັ້ນຫນຽວເສັ້ນດຽວໃນຊິ້ນສ່ວນທີ່ໃສ່ຫຼັງຈາກເວລາສັ້ນໆ.ເນື່ອງຈາກວ່າຜະລິດຕະພັນສາມາດໄດ້ຮັບການສ້າງຕັ້ງຂຶ້ນພຽງແຕ່ຈາກການ annealing ເຊິ່ງກັນແລະກັນຂອງຊິ້ນສະແດງກິ່ງງ່າທີ່ກະກຽມແລະ vector, ວິທີການນີ້ແມ່ນໄວແລະປະສິດທິພາບຫຼາຍ, ແລະມັນແມ່ນການຊີ້ນໍາ cloning.
ທິດທາງ TA clone ເພີ່ມຫາງ
TA cloning ບໍ່ສາມາດແນເປົ້າໃສ່ຊິ້ນສ່ວນເຂົ້າໄປໃນ vector ໄດ້, ສະນັ້ນຕໍ່ມາ Invitrogen ແນະນໍາ vector ທີ່ສາມາດເປົ້າຫມາຍ cloning, ເຊິ່ງປະກອບດ້ວຍສີ່ຖານທີ່ໂດດເດັ່ນ GTGGS ຢູ່ປາຍຫນຶ່ງ.ດັ່ງນັ້ນ, ໃນການອອກແບບ primers PCR, ລໍາດັບເສີມຄວນໄດ້ຮັບການເພີ່ມຕາມຄວາມເຫມາະສົມ, ດັ່ງນັ້ນຊິ້ນສ່ວນສາມາດ "ຮັດກຸມ".
ຖ້າທ່ານຂາດເວລາ, ທ່ານສາມາດທົດລອງການສັງເຄາະໂດຍກົງ, ສົມທົບ gene ກັບ vector, ເຊິ່ງພວກເຮົາເອີ້ນວ່າການສັງເຄາະ ET ໃນ musecularists.
D. In-Fusion ວິທີການ cloning
ບໍ່ຕ້ອງການ ligase, ບໍ່ຕ້ອງການຕິກິຣິຍາຍາວ.ຕາບໃດທີ່ລໍາດັບຢູ່ທັງສອງສົ້ນຂອງ vector linearized ຖືກນໍາສະເຫນີໃນການອອກແບບ primers, ຫຼັງຈາກນັ້ນຜະລິດຕະພັນ PCR ແລະ vector linearized ໄດ້ຖືກເພີ່ມເຂົ້າໃນການແກ້ໄຂ enzyme in-fusion ທີ່ມີ BSA ແລະວາງໄວ້ໃນອຸນຫະພູມຫ້ອງສໍາລັບເຄິ່ງຊົ່ວໂມງ, ການຫັນປ່ຽນສາມາດປະຕິບັດໄດ້.ວິທີການນີ້ແມ່ນເຫມາະສົມໂດຍສະເພາະສໍາລັບການປ່ຽນປະລິມານຂະຫນາດໃຫຍ່.
10. ປະສົມ primer
ບາງຄັ້ງ, ພຽງແຕ່ຂໍ້ມູນລໍາດັບຈໍາກັດແມ່ນເປັນທີ່ຮູ້ຈັກກ່ຽວກັບການອອກແບບ primer.ຕົວຢ່າງ, ຖ້າພຽງແຕ່ລໍາດັບອາຊິດ amino ເປັນທີ່ຮູ້ຈັກ, primer ປະສົມປະສານສາມາດອອກແບບໄດ້.primer ການລວມຕົວແມ່ນສ່ວນປະສົມຂອງລໍາດັບທີ່ແຕກຕ່າງກັນທີ່ເປັນຕົວແທນຂອງຄວາມເປັນໄປໄດ້ພື້ນຖານທີ່ແຕກຕ່າງກັນທັງຫມົດທີ່ເຂົ້າລະຫັດອາຊິດ amino ດຽວ.ເພື່ອເພີ່ມຄວາມສະເພາະ, ທ່ານສາມາດອ້າງອີງໃສ່ຕາຕະລາງການນໍາໃຊ້ codon ເພື່ອຫຼຸດຜ່ອນການຊ້ອນກັນໂດຍອີງຕາມຄວາມມັກການນໍາໃຊ້ພື້ນຖານຂອງອົງການຈັດຕັ້ງທີ່ແຕກຕ່າງກັນ.Hypoxanthine ສາມາດຖືກຈັບຄູ່ກັບຖານທັງຫມົດເພື່ອຫຼຸດຜ່ອນອຸນຫະພູມການຫມູນວຽນຂອງ primer.ຢ່າໃຊ້ຖານທີ່ຊ້ອນກັນຢູ່ປາຍ 3′ ຂອງ primer ເພາະວ່າການເຊື່ອມຂອງຖານ 3 ສຸດທ້າຍຢູ່ທີ່ປາຍ 3′ ແມ່ນພຽງພໍທີ່ຈະເລີ່ມຕົ້ນ PCR ຢູ່ໃນບ່ອນທີ່ບໍ່ຖືກຕ້ອງ.ຄວາມເຂັ້ມຂຸ້ນຂອງ primer ທີ່ສູງຂຶ້ນ (1μM ຫາ 3μM) ແມ່ນຖືກນໍາໃຊ້ເພາະວ່າ primers ໃນສ່ວນຜະສົມຜະສານຊ້ອນກັນຫຼາຍແມ່ນບໍ່ສະເພາະກັບແມ່ແບບເປົ້າຫມາຍ.
ວັດຖຸດິບ PCRການຄວບຄຸມ
1. ປະລິມານ primer
ຄວາມເຂັ້ມຂົ້ນຂອງ primer ແຕ່ລະຄົນແມ່ນ 0.1 ~ 1umol ຫຼື 10 ~ 100pmol.ມັນດີກວ່າທີ່ຈະຜະລິດຜົນໄດ້ຮັບທີ່ຕ້ອງການດ້ວຍປະລິມານຕ່ໍາສຸດຂອງ primer.ຄວາມເຂັ້ມຂຸ້ນສູງຂອງ primer ຈະເຮັດໃຫ້ເກີດການຂະຫຍາຍທີ່ບໍ່ກົງກັນແລະບໍ່ສະເພາະ, ແລະເພີ່ມໂອກາດຂອງການສ້າງ dimers ລະຫວ່າງ primers.
2. ຄວາມເຂັ້ມຂຸ້ນຂອງ primer
ຄວາມເຂັ້ມຂົ້ນຂອງ primers ມີຜົນກະທົບສະເພາະ.ຄວາມເຂັ້ມຂຸ້ນຂອງ primer ທີ່ດີທີ່ສຸດແມ່ນໂດຍທົ່ວໄປລະຫວ່າງ 0.1 ແລະ 0.5μM.ຄວາມເຂັ້ມຂຸ້ນຂອງ primer ທີ່ສູງຂຶ້ນນໍາໄປສູ່ການຂະຫຍາຍຜະລິດຕະພັນທີ່ບໍ່ສະເພາະ.
3. ອຸນຫະພູມ Annealing ຂອງ primer
ຕົວກໍານົດການທີ່ສໍາຄັນອີກອັນຫນຶ່ງສໍາລັບ primers ແມ່ນອຸນຫະພູມ melting (Tm).ນີ້ແມ່ນອຸນຫະພູມໃນເວລາທີ່ 50% ຂອງ primers ແລະລໍາດັບເສີມແມ່ນເປັນຕົວແທນເປັນໂມເລກຸນ DNA ຄູ່.Tm ແມ່ນຈໍາເປັນເພື່ອກໍານົດອຸນຫະພູມການຫມູນວຽນ PCR.ໂດຍຫລັກການແລ້ວ, ອຸນຫະພູມການຫມຸນແມ່ນຕ່ໍາພຽງພໍທີ່ຈະຮັບປະກັນການຫມູນວຽນທີ່ມີປະສິດທິພາບຂອງ primers ກັບລໍາດັບເປົ້າຫມາຍ, ແຕ່ສູງພຽງພໍທີ່ຈະຫຼຸດຜ່ອນການຜູກມັດທີ່ບໍ່ສະເພາະ.ອຸນຫະພູມ annealing ສົມເຫດສົມຜົນຈາກ 55 ℃ເຖິງ 70 ℃.ອຸນຫະພູມ Annealing ໂດຍທົ່ວໄປແມ່ນກໍານົດ 5 ℃ຕ່ໍາກ່ວາ Tm ຂອງ primer.
ມີຫຼາຍສູດສໍາລັບການຕັ້ງຄ່າ Tm, ເຊິ່ງແຕກຕ່າງກັນຢ່າງຫຼວງຫຼາຍຂຶ້ນຢູ່ກັບສູດທີ່ໃຊ້ແລະລໍາດັບຂອງ primers.ເນື່ອງຈາກວ່າສູດສ່ວນໃຫຍ່ໃຫ້ຄ່າ Tm ຄາດຄະເນ, ອຸນຫະພູມການຫມູນວຽນທັງຫມົດແມ່ນພຽງແຕ່ຈຸດເລີ່ມຕົ້ນເທົ່ານັ້ນ.ຄວາມສະເພາະສາມາດປັບປຸງໄດ້ໂດຍການວິເຄາະປະຕິກິລິຢາຫຼາຍອັນທີ່ເພີ່ມອຸນຫະພູມການຫມູນວຽນຢ່າງກ້າວກະໂດດ.ເລີ່ມຕົ້ນຕ່ໍາກວ່າ Tm-5 ℃, ແລະຄ່ອຍໆເພີ່ມອຸນຫະພູມ annealing ເພີ່ມຂຶ້ນເປັນ 2 ℃.ອຸນຫະພູມ annealing ສູງຂຶ້ນຈະຫຼຸດຜ່ອນການສ້າງຕັ້ງຂອງ primer dimers ແລະຜະລິດຕະພັນທີ່ບໍ່ແມ່ນສະເພາະ.ສໍາລັບຜົນໄດ້ຮັບທີ່ດີທີ່ສຸດ, ສອງ primers ຄວນມີຄ່າ Tm ປະມານ.ຖ້າຄວາມແຕກຕ່າງຂອງ Tm ຂອງຄູ່ primer ແມ່ນຫຼາຍກ່ວາ 5 ℃, primers ຈະສະແດງໃຫ້ເຫັນການເລີ່ມຕົ້ນທີ່ບໍ່ຖືກຕ້ອງທີ່ສໍາຄັນໂດຍການນໍາໃຊ້ອຸນຫະພູມຕ່ໍາ annealing ໃນວົງຈອນ.ຖ້າສອງ primers Tm ແຕກຕ່າງກັນ, ກໍານົດອຸນຫະພູມ annealing ເປັນ 5 ℃ຕ່ໍາກວ່າ Tm ຕ່ໍາສຸດ.ອີກທາງເລືອກ, ເພື່ອເພີ່ມຄວາມສະເພາະ, ຫ້າຮອບສາມາດປະຕິບັດຄັ້ງທໍາອິດໃນອຸນຫະພູມ annealing ອອກແບບສໍາລັບ Tm ສູງຂຶ້ນ, ຕິດຕາມດ້ວຍຮອບວຽນທີ່ຍັງເຫຼືອຢູ່ໃນອຸນຫະພູມ annealing ອອກແບບສໍາລັບ Tm ຕ່ໍາ.ນີ້ອະນຸຍາດໃຫ້ສໍາເນົາບາງສ່ວນຂອງແມ່ແບບປາຍທາງທີ່ຈະໄດ້ຮັບພາຍໃຕ້ເງື່ອນໄຂທີ່ເຄັ່ງຄັດ.
4. ຄວາມບໍລິສຸດຂອງ primer ແລະຄວາມຫມັ້ນຄົງ
ຄວາມບໍລິສຸດມາດຕະຖານຂອງ primers ທີ່ກໍາຫນົດເອງແມ່ນພຽງພໍສໍາລັບຄໍາຮ້ອງສະຫມັກ PCR ສ່ວນໃຫຍ່.ການໂຍກຍ້າຍຂອງກຸ່ມ benzoyl ແລະ isobutylyl ໂດຍ desalting ແມ່ນຫນ້ອຍທີ່ສຸດແລະດັ່ງນັ້ນຈຶ່ງບໍ່ແຊກແຊງກັບ PCR.ຄໍາຮ້ອງສະຫມັກຈໍານວນຫນຶ່ງຮຽກຮ້ອງໃຫ້ມີການຊໍາລະລ້າງເພື່ອກໍາຈັດລໍາດັບທີ່ບໍ່ມີຄວາມຍາວເຕັມໃນຂະບວນການສັງເຄາະ.ລໍາດັບທີ່ຖືກຕັດອອກເຫຼົ່ານີ້ເກີດຂື້ນຍ້ອນວ່າປະສິດທິພາບຂອງເຄມີສັງເຄາະ DNA ບໍ່ແມ່ນ 100%.ນີ້ແມ່ນຂະບວນການເປັນວົງທີ່ໃຊ້ຕິກິຣິຍາທາງເຄມີຊ້ໍາເນື່ອງຈາກວ່າແຕ່ລະຖານໄດ້ຖືກເພີ່ມເພື່ອເຮັດໃຫ້ DNA ຈາກ 3′ ຫາ 5′.ທ່ານສາມາດລົ້ມເຫລວໃນທັງສອງວົງຈອນ.primers ຍາວ, ໂດຍສະເພາະແມ່ນພື້ນຖານທີ່ໃຫຍ່ກວ່າ 50, ມີອັດຕາສ່ວນຂະຫນາດໃຫຍ່ຂອງລໍາດັບທີ່ຖືກຕັດອອກແລະອາດຈະຕ້ອງການການເຮັດຄວາມສະອາດ.
ຜົນຜະລິດຂອງ primers ໄດ້ຮັບຜົນກະທົບຈາກປະສິດທິພາບຂອງເຄມີສັງເຄາະແລະວິທີການ purification.ບໍລິສັດຢາຊີວະພາບ, ເຊັ່ນ Cytology ແລະ Shengong, ທັງຫມົດນໍາໃຊ້ຫນ່ວຍງານ OD ຕ່ໍາສຸດເພື່ອຮັບປະກັນຜົນຜະລິດທັງຫມົດຂອງ oligonucleoside.primers ແບບກໍາຫນົດເອງຖືກຈັດສົ່ງໃນຮູບແບບຝຸ່ນແຫ້ງ.ມັນດີທີ່ສຸດທີ່ຈະແກ້ໄຂ primers ໃນ TE ເພື່ອຄວາມເຂັ້ມຂົ້ນສຸດທ້າຍແມ່ນ 100μM.TE ແມ່ນດີກວ່ານ້ໍາ deionized ເພາະວ່າ pH ຂອງນ້ໍາມັກຈະເປັນກົດແລະຈະເຮັດໃຫ້ hydrolysis ຂອງ oligonucleosides.
ຄວາມຫມັ້ນຄົງຂອງ primers ແມ່ນຂຶ້ນກັບເງື່ອນໄຂການເກັບຮັກສາ.ຝຸ່ນແຫ້ງແລະ primers ທີ່ລະລາຍຄວນໄດ້ຮັບການເກັບຮັກສາໄວ້ທີ່ -20 ℃.primers ທີ່ລະລາຍໃນ TE ທີ່ມີຄວາມເຂັ້ມຂຸ້ນສູງກວ່າ 10μM ສາມາດເກັບຮັກສາໄວ້ຢ່າງຫມັ້ນຄົງຢູ່ທີ່ -20 ℃ສໍາລັບ 6 ເດືອນ, ແຕ່ສາມາດເກັບຮັກສາໄວ້ໃນອຸນຫະພູມຫ້ອງ (15 ℃ເຖິງ 30 ℃) ຫນ້ອຍກວ່າ 1 ອາທິດ.ຝຸ່ນ primers ແຫ້ງສາມາດເກັບຮັກສາໄວ້ຢູ່ທີ່ -20 C ເປັນເວລາຢ່າງຫນ້ອຍ 1 ປີແລະໃນອຸນຫະພູມຫ້ອງ (15 C ຫາ 30 C) ດົນເຖິງ 2 ເດືອນ.
5. Enzymes ແລະຄວາມເຂັ້ມຂຸ້ນຂອງພວກມັນ
ໃນປັດຈຸບັນ, Taq DNA polymerase ທີ່ໃຊ້ໂດຍພື້ນຖານແລ້ວແມ່ນ enzyme ວິສະວະກໍາ gene ທີ່ສັງເຄາະໂດຍເຊື້ອແບັກທີເຣັຍ coliform.ປະລິມານຂອງເອນໄຊທີ່ຈໍາເປັນເພື່ອກະຕຸ້ນປະຕິກິລິຍາ PCR ປົກກະຕິແມ່ນປະມານ 2.5U (ຫມາຍເຖິງປະລິມານຕິກິຣິຍາທັງຫມົດ 100ul).ຖ້າຄວາມເຂັ້ມຂົ້ນສູງເກີນໄປ, ມັນສາມາດນໍາໄປສູ່ການຂະຫຍາຍທີ່ບໍ່ສະເພາະ;ຖ້າຄວາມເຂັ້ມຂົ້ນຕໍ່າເກີນໄປ, ປະລິມານຂອງຜະລິດຕະພັນສັງເຄາະຈະຫຼຸດລົງ.
6. ຄຸນນະພາບແລະຄວາມເຂັ້ມຂົ້ນຂອງ dNTP
ຄຸນນະພາບຂອງ dNTP ແມ່ນກ່ຽວຂ້ອງຢ່າງໃກ້ຊິດກັບຄວາມເຂັ້ມຂົ້ນແລະປະສິດທິພາບຂອງການຂະຫຍາຍ PCR.ຜົງ dNTP ເປັນເມັດ, ແລະຄວາມປ່ຽນແປງຂອງມັນສູນເສຍການເຄື່ອນໄຫວທາງຊີວະພາບຂອງມັນຖ້າມັນຖືກເກັບໄວ້ບໍ່ຖືກຕ້ອງ.ການແກ້ໄຂ dNTP ເປັນກົດ, ແລະຄວນຈະຖືກນໍາໃຊ້ໃນຄວາມເຂັ້ມຂົ້ນສູງ, ມີການແກ້ໄຂ 1M NaOH ຫຼື 1M Tris.HCL buffer ເພື່ອປັບ PH ຂອງຕົນເປັນ 7.0 ~ 7.5, ຈໍານວນຂະຫນາດນ້ອຍຂອງການຫຸ້ມຫໍ່ຍ່ອຍ, ການເກັບຮັກສາ frozen ຢູ່ -20 ℃.ການແຊ່ແຂງຫຼາຍຄັ້ງຈະເຮັດໃຫ້ dNTP ຫຼຸດລົງ.ໃນປະຕິກິລິຍາ PCR, dNTP ຄວນຈະເປັນ 50 ~ 200umol / L.ໂດຍສະເພາະ, ຄວນເອົາໃຈໃສ່ກັບຄວາມເຂັ້ມຂົ້ນຂອງສີ່ DNTPS ຄວນມີຄວາມເທົ່າທຽມກັນ (ການກະກຽມ mole ເທົ່າທຽມກັນ).ຖ້າຄວາມເຂັ້ມຂຸ້ນຂອງອັນໃດອັນໜຶ່ງແຕກຕ່າງຈາກອັນອື່ນ (ສູງກວ່າ ຫຼືຕ່ຳ), ຄວາມບໍ່ສອດຄ່ອງຈະເກີດ.ຄວາມເຂັ້ມຂົ້ນຕ່ໍາເກີນໄປຈະຫຼຸດລົງຜົນຜະລິດຂອງຜະລິດຕະພັນ PCR.dNTP ສາມາດສົມທົບກັບ Mg2+ ແລະຫຼຸດຜ່ອນຄວາມເຂັ້ມຂົ້ນຂອງ Mg2+ ຟຣີ.
7. ແມ່ແບບ (ເຊື້ອພັນເປົ້າໝາຍ) ອາຊິດນິວຄລີອິກ
ປະລິມານແລະລະດັບການຊໍາລະຂອງອາຊິດ nucleic ແມ່ແບບແມ່ນຫນຶ່ງໃນການເຊື່ອມໂຍງທີ່ສໍາຄັນສໍາລັບຄວາມສໍາເລັດຫຼືຄວາມລົ້ມເຫຼວຂອງ PCR.ວິທີການຊໍາລະລ້າງ DNA ແບບດັ້ງເດີມມັກຈະໃຊ້ SDS ແລະ protease K ເພື່ອຍ່ອຍອາຫານແລະກໍາຈັດຕົວຢ່າງ.ຫນ້າທີ່ຕົ້ນຕໍຂອງ SDS ແມ່ນ: ການລະລາຍ lipids ແລະທາດໂປຼຕີນໃນເຍື່ອຂອງຈຸລັງ, ດັ່ງນັ້ນການທໍາລາຍເຍື່ອຈຸລັງໂດຍການລະລາຍໂປຣຕີນຂອງເຍື່ອ, ແລະ dissociate ທາດໂປຼຕີນຈາກນິວເຄລຍໃນຈຸລັງ, SDS ຍັງສາມາດສົມທົບກັບທາດໂປຼຕີນແລະ precipitate;Protease K ສາມາດ hydrolyze ແລະຍ່ອຍທາດໂປຼຕີນ, ໂດຍສະເພາະແມ່ນ histones ທີ່ຜູກມັດກັບ DNA, ແລະຫຼັງຈາກນັ້ນນໍາໃຊ້ສານລະລາຍອິນຊີ phenol ແລະ chloroform ເພື່ອສະກັດທາດໂປຼຕີນແລະອົງປະກອບຂອງເຊນອື່ນໆ, ແລະນໍາໃຊ້ເອທານອນຫຼື isopropyl ເຫຼົ້າເພື່ອ precipitate ອາຊິດ nucleic.ອາຊິດນິວຄລີອິກທີ່ສະກັດອອກມາສາມາດຖືກນໍາໃຊ້ເປັນແມ່ແບບສໍາລັບປະຕິກິລິຍາ PCR.ສໍາລັບຕົວຢ່າງການກວດສອບທາງດ້ານການຊ່ວຍທົ່ວໄປ, ວິທີການໄວແລະງ່າຍດາຍສາມາດຖືກນໍາໃຊ້ເພື່ອລະລາຍຈຸລັງ, lysate ເຊື້ອພະຍາດ, ຍ່ອຍອາຫານແລະເອົາທາດໂປຼຕີນຈາກໂຄໂມໂຊມເພື່ອພັນທຸກໍາເປົ້າຫມາຍຟຣີ, ແລະນໍາໃຊ້ໂດຍກົງສໍາລັບການຂະຫຍາຍ PCR.ການສະກັດເອົາແມ່ແບບ RNA ປົກກະຕິແລ້ວໃຊ້ວິທີ guanidine isothiocyanate ຫຼື protease K ເພື່ອປ້ອງກັນ RNase ຈາກການທໍາລາຍ RNA.
ຄວາມເຂັ້ມຂຸ້ນ 8.Mg2+
Mg2+ ມີຜົນກະທົບຢ່າງຫຼວງຫຼາຍຕໍ່ຄວາມສະເພາະແລະຜົນຜະລິດຂອງການຂະຫຍາຍ PCR.ໂດຍທົ່ວໄປປະຕິກິລິຍາ PCR, ເມື່ອຄວາມເຂັ້ມຂົ້ນຂອງ dNTP ຕ່າງໆແມ່ນ 200umol / L, ຄວາມເຂັ້ມຂົ້ນທີ່ເຫມາະສົມຂອງ Mg2+ ແມ່ນ 1.5 ~ 2.0mmol / L.ຄວາມເຂັ້ມຂົ້ນຂອງ Mg2+ ແມ່ນສູງເກີນໄປ, ຄວາມສະເພາະຂອງຕິກິຣິຍາຫຼຸດລົງ, ການຂະຫຍາຍທີ່ບໍ່ສະເພາະເກີດຂື້ນ, ຄວາມເຂັ້ມຂົ້ນຕ່ໍາເກີນໄປຈະຫຼຸດລົງກິດຈະກໍາຂອງ Taq DNA polymerase, ເຊິ່ງກໍ່ໃຫ້ເກີດການຫຼຸດຜ່ອນການປະຕິກິລິຍາຂອງຜະລິດຕະພັນ.
ions ແມກນີຊຽມມີຜົນກະທົບຫຼາຍດ້ານຂອງ PCR, ເຊັ່ນ: ກິດຈະກໍາ DNA polymerase, ເຊິ່ງຜົນກະທົບຕໍ່ຜົນຜະລິດ;ຕົວຢ່າງອື່ນແມ່ນ primer annealing, ເຊິ່ງມີຜົນກະທົບສະເພາະ.dNTP ແລະແມ່ແບບຜູກມັດກັບ magnesium ion, ຫຼຸດຜ່ອນຈໍານວນ magnesium ion ຟຣີທີ່ຕ້ອງການສໍາລັບກິດຈະກໍາ enzyme.ຄວາມເຂັ້ມຂຸ້ນຂອງ magnesium ion ທີ່ດີທີ່ສຸດແມ່ນແຕກຕ່າງກັນສໍາລັບຄູ່ primer ແລະແມ່ແບບທີ່ແຕກຕ່າງກັນ, ແຕ່ຄວາມເຂັ້ມຂົ້ນຂອງ PCR ປົກກະຕິທີ່ມີ 200μM dNTP ແມ່ນ 1.5mM (ຫມາຍເຫດ: ສໍາລັບ PCR ປະລິມານທີ່ແທ້ຈິງ, ໃຫ້ໃຊ້ 3 ຫາ 5mM magnesium ion solution ທີ່ມີ probe fluorescent).ຄວາມເຂັ້ມຂຸ້ນທີ່ສູງຂຶ້ນຂອງ ions magnesium ຟຣີເພີ່ມຜົນຜະລິດ, ແຕ່ຍັງເພີ່ມການຂະຫຍາຍ nonspecific ແລະຫຼຸດລົງຄວາມຊື່ສັດ.ເພື່ອກໍານົດຄວາມເຂັ້ມຂົ້ນທີ່ດີທີ່ສຸດ, ການ titration magnesium ion ໄດ້ຖືກປະຕິບັດໃນເພີ່ມຂຶ້ນ 0.5mM ຈາກ 1mM ຫາ 3mM.ເພື່ອຫຼຸດຜ່ອນການເພິ່ງພາອາໄສການເພີ່ມປະສິດທິພາບຂອງ magnesium ion, Platinum Taq DNA polymerase ສາມາດນໍາໃຊ້ໄດ້.Platinum Taq DNA polymerase ສາມາດຮັກສາການທໍາງານຂອງຄວາມເຂັ້ມຂຸ້ນຂອງ magnesium ion ທີ່ກວ້າງຂວາງກວ່າ Taq DNA polymerase ແລະດັ່ງນັ້ນຈຶ່ງຕ້ອງການການເພີ່ມປະສິດທິພາບຫນ້ອຍ.
9. Pcr-promoting additives
ການເພີ່ມປະສິດທິພາບຂອງອຸນຫະພູມ annealing, ການອອກແບບ primer, ແລະຄວາມເຂັ້ມຂົ້ນຂອງ magnesium ion ແມ່ນພຽງພໍສໍາລັບການຂະຫຍາຍທີ່ສະເພາະຂອງແມ່ແບບສ່ວນໃຫຍ່;ຢ່າງໃດກໍຕາມ, ບາງແມ່ແບບ, ລວມທັງຜູ້ທີ່ມີເນື້ອໃນ GC ສູງ, ຮຽກຮ້ອງໃຫ້ມີມາດຕະການເພີ່ມເຕີມ.ສານເຕີມແຕ່ງທີ່ມີຜົນກະທົບຕໍ່ອຸນຫະພູມການລະລາຍຂອງ DNA ໃຫ້ວິທີການອື່ນເພື່ອປັບປຸງຄວາມສະເພາະຂອງຜະລິດຕະພັນແລະຜົນຜະລິດ.ຕ້ອງມີການປ່ຽນຕົວແບບເຕັມຮູບແບບເພື່ອໃຫ້ໄດ້ຜົນທີ່ດີທີ່ສຸດ.
ນອກຈາກນັ້ນ, ໂຄງສ້າງຂັ້ນສອງປ້ອງກັນການຜູກມັດ primer ແລະການຂະຫຍາຍ enzyme.
ສານເສີມ PCR, ລວມທັງ formamide, DMSO, glycerin, betaine, ແລະ PCRx Enhancer Solution, ເສີມຂະຫຍາຍການຂະຫຍາຍ.ກົນໄກທີ່ເປັນໄປໄດ້ຂອງພວກເຂົາແມ່ນເພື່ອຫຼຸດຜ່ອນອຸນຫະພູມການລະລາຍ, ດັ່ງນັ້ນການຊ່ວຍເຫຼືອການຫມູນວຽນຂອງ primers ແລະຊ່ວຍຂະຫຍາຍ DNA polymerase ຜ່ານພາກພື້ນໂຄງສ້າງຮອງ.PCRx Solution ມີຂໍ້ດີອື່ນໆ.ການເພີ່ມປະສິດທິພາບຂອງ magnesium ion ໜ້ອຍທີ່ສຸດແມ່ນຕ້ອງການເມື່ອໃຊ້ກັບ Platinum Taq DNA polymerase ແລະ Platinum Pfx DNA polymerase.ດັ່ງນັ້ນ, ເທກນິກ Platinum ແມ່ນປະສົມປະສານກັບສານເສີມເພື່ອເພີ່ມຄວາມສະເພາະໃນຂະນະທີ່ການຫຼຸດຜ່ອນການເພິ່ງພາອາໄສຂອງວິທີການທີສາມ, ການເພີ່ມປະສິດທິພາບຂອງ magnesium ion.ສໍາລັບຜົນໄດ້ຮັບທີ່ດີທີ່ສຸດ, ຄວາມເຂັ້ມຂົ້ນຂອງສານເຕີມແຕ່ງຄວນໄດ້ຮັບການເພີ່ມປະສິດທິພາບ, ໂດຍສະເພາະ DMSO, formamide, ແລະ glycerol, ເຊິ່ງ inhibit Taq DNA polymerase.

10. ເລີ່ມຕົ້ນຮ້ອນ
PCR ເລີ່ມຕົ້ນຮ້ອນແມ່ນຫນຶ່ງໃນວິທີການທີ່ສໍາຄັນທີ່ສຸດເພື່ອປັບປຸງຄວາມສະເພາະຂອງ PCR ນອກເຫນືອຈາກການອອກແບບ primer ທີ່ດີ.ເຖິງແມ່ນວ່າອຸນຫະພູມການຍືດຕົວທີ່ດີທີ່ສຸດຂອງ Taq DNA polymerase ແມ່ນ 72 ℃, polymerase ຍັງຄົງມີການເຄື່ອນໄຫວຢູ່ໃນອຸນຫະພູມຫ້ອງ.ດັ່ງນັ້ນ, ຜະລິດຕະພັນທີ່ບໍ່ສະເພາະແມ່ນຜະລິດໃນເວລາທີ່ອຸນຫະພູມຖືຕ່ໍາກວ່າອຸນຫະພູມ annealing ໃນລະຫວ່າງການກະກຽມປະຕິກິລິຍາ PCR ແລະໃນຕອນຕົ້ນຂອງວົງຈອນຄວາມຮ້ອນ.ເມື່ອຖືກສ້າງຕັ້ງຂຶ້ນ, ຜະລິດຕະພັນທີ່ບໍ່ສະເພາະເຫຼົ່ານີ້ຖືກຂະຫຍາຍອອກຢ່າງມີປະສິດທິພາບ.PCR ເລີ່ມຕົ້ນທີ່ມີປະສິດຕິຜົນໂດຍສະເພາະເມື່ອສະຖານທີ່ທີ່ໃຊ້ສໍາລັບການອອກແບບ primer ຖືກຈໍາກັດໂດຍສະຖານທີ່ຂອງອົງປະກອບທາງພັນທຸກໍາ, ເຊັ່ນ: ການກາຍພັນຂອງສະຖານທີ່, ການສະແດງອອກ, ການສະແດງອອກ, ຫຼືການກໍ່ສ້າງແລະການຫມູນໃຊ້ຂອງອົງປະກອບທາງພັນທຸກໍາທີ່ໃຊ້ສໍາລັບວິສະວະກໍາ DNA.
ວິທີການທົ່ວໄປເພື່ອຈໍາກັດກິດຈະກໍາຂອງ Taq DNA polymerase ແມ່ນການກະກຽມການແກ້ໄຂປະຕິກິລິຍາ PCR ເທິງກ້ອນແລະວາງໄວ້ໃນອຸປະກອນ PCR ທີ່ມີຄວາມຮ້ອນກ່ອນ.ວິທີການນີ້ແມ່ນງ່າຍດາຍແລະລາຄາບໍ່ແພງ, ແຕ່ມັນບໍ່ສໍາເລັດກິດຈະກໍາຂອງ enzyme ແລະດັ່ງນັ້ນຈິ່ງບໍ່ໄດ້ລົບລ້າງການຂະຫຍາຍຂອງຜະລິດຕະພັນທີ່ບໍ່ສະເພາະ.
ການປະສົມຄວາມຮ້ອນເຮັດໃຫ້ການສັງເຄາະ DNA ຊັກຊ້າໂດຍການຍັບຍັ້ງອົງປະກອບທີ່ສໍາຄັນຈົນກ່ວາອຸປະກອນ PCR ຮອດອຸນຫະພູມທີ່ບໍ່ເປັນທໍາມະຊາດ.ວິທີການເລີ່ມຕົ້ນຄວາມຮ້ອນດ້ວຍມືສ່ວນໃຫຍ່, ລວມທັງການຊັກຊ້າຂອງການເພີ່ມ Taq DNA polymerase, ແມ່ນມີຄວາມຫຍຸ້ງຍາກ, ໂດຍສະເພາະສໍາລັບຄໍາຮ້ອງສະຫມັກທີ່ມີຄວາມໄວສູງ.ວິທີການ priming ຄວາມຮ້ອນອື່ນໆໃຊ້ wax ໄສ້ເພື່ອປະກອບອົງປະກອບທີ່ສໍາຄັນ, ລວມທັງ magnesium ions ຫຼື enzymes, ຫຼືເພື່ອແຍກອົງປະກອບ reactive, ເຊັ່ນ: ແມ່ແບບແລະ buffers.ໃນລະຫວ່າງວົງຈອນຄວາມຮ້ອນ, ອົງປະກອບຕ່າງໆຖືກປ່ອຍອອກມາແລະປະສົມເຂົ້າກັນໃນຂະນະທີ່ຂີ້ເຜີ້ງ melts.ເຊັ່ນດຽວກັນກັບວິທີການເລີ່ມຕົ້ນຮ້ອນດ້ວຍມື, ວິທີການໄສ້ຂີ້ເຜີ້ງແມ່ນຫຍຸ້ງຍາກແລະມີຄວາມສ່ຽງຕໍ່ການປົນເປື້ອນແລະບໍ່ເຫມາະສົມສໍາລັບຄໍາຮ້ອງສະຫມັກທີ່ມີຄວາມໄວສູງ.
Platinum DNA polymerase ແມ່ນສະດວກແລະປະສິດທິພາບສໍາລັບ PCR ເລີ່ມຕົ້ນຮ້ອນອັດຕະໂນມັດ.Platinum Taq DNA polymerase ປະກອບດ້ວຍ Taq DNA polymerase ທີ່ປະສົມປະສານກັບພູມຕ້ານທານ monoclonal ຕໍ່ Taq DNA polymerase.ພູມຕ້ານທານແມ່ນສ້າງຂື້ນໂດຍ PCR ເພື່ອຍັບຍັ້ງກິດຈະກໍາຂອງເອນໄຊໃນລະຫວ່າງການຮັກສາອຸນຫະພູມເປັນເວລາດົນນານ.Taq DNA polymerase ໄດ້ຖືກປ່ອຍອອກມາໃນຕິກິຣິຍາໃນລະຫວ່າງການ insulation 94 ℃ຂອງຂັ້ນຕອນ denaturation, ການຟື້ນຟູກິດຈະກໍາ polymerase ຢ່າງເຕັມທີ່.ໃນທາງກົງກັນຂ້າມກັບ Taq DNA polymerase ທີ່ຖືກດັດແປງທາງເຄມີສໍາລັບການເລີ່ມຕົ້ນຄວາມຮ້ອນ, ເອນໄຊ Platinum ບໍ່ຈໍາເປັນຕ້ອງມີ insulation ທີ່ຍາວນານຢູ່ທີ່ 94 ℃ (10 ຫາ 15 ນາທີ) ເພື່ອກະຕຸ້ນໂພລີເມີເຣສ.ດ້ວຍ PlatinumTaq DNA polymerase, 90% ຂອງກິດຈະກໍາ Taq DNA polymerase ໄດ້ຖືກຟື້ນຟູຄືນມາພາຍຫຼັງ 2 ນາທີຢູ່ທີ່ 94 ℃.

11. Nest-PCR
ການຂະຫຍາຍຮອບຢ່າງຕໍ່ເນື່ອງໂດຍໃຊ້ primers ຊ້ອນສາມາດປັບປຸງສະເພາະແລະຄວາມອ່ອນໄຫວ.ຮອບທໍາອິດແມ່ນການຂະຫຍາຍມາດຕະຖານ 15 ຫາ 20 ຮອບ.ສ່ວນຂະຫນາດນ້ອຍຂອງຜະລິດຕະພັນຂະຫຍາຍໃຫຍ່ຂື້ນໃນເບື້ອງຕົ້ນໄດ້ຖືກ diluted 100 ຫາ 1000 ເທື່ອແລະເພີ່ມເຂົ້າໄປໃນຮອບທີສອງຂອງການຂະຫຍາຍສໍາລັບ 15 ຫາ 20 ຮອບ.ອີກທາງເລືອກ, ຜະລິດຕະພັນທີ່ຂະຫຍາຍໃຫຍ່ຂື້ນໃນເບື້ອງຕົ້ນສາມາດຖືກຂະຫນາດໂດຍການເຮັດຄວາມສະອາດ gel.primer ຮັງຖືກນໍາໃຊ້ໃນຮອບທີສອງຂອງການຂະຫຍາຍ, ເຊິ່ງສາມາດຜູກມັດກັບລໍາດັບເປົ້າຫມາຍພາຍໃນ primer ທໍາອິດ.ການນໍາໃຊ້ PCR ທີ່ຖືກວາງໄວ້ຊ່ວຍຫຼຸດຜ່ອນຄວາມເປັນໄປໄດ້ຂອງການຂະຫຍາຍສະຖານທີ່ເປົ້າຫມາຍຫຼາຍເພາະວ່າມີລໍາດັບເປົ້າຫມາຍຈໍານວນຫນ້ອຍທີ່ປະສົມປະສານກັບທັງສອງຊຸດຂອງ primers.ຈໍານວນທັງຫມົດຂອງວົງຈອນດຽວກັນ (30 ຫາ 40) ທີ່ມີ primers ດຽວກັນຂະຫຍາຍສະຖານທີ່ທີ່ບໍ່ຈໍາເປັນ.Nested PCR ເພີ່ມຄວາມອ່ອນໄຫວຂອງລໍາດັບເປົ້າຫມາຍທີ່ຈໍາກັດ (ຕົວຢ່າງ, mrnas ທີ່ຫາຍາກ) ແລະປັບປຸງຄວາມສະເພາະຂອງ PCRS ຍາກ (ເຊັ່ນ: 5′ RACE).
12. ລົງ PCR
Descending PCR ປັບປຸງຄວາມສະເພາະໂດຍການນໍາໃຊ້ເງື່ອນໄຂການບີບອັດແຫນ້ນສໍາລັບສອງສາມຮອບທໍາອິດຂອງ PCR.ວົງຈອນເລີ່ມຕົ້ນຢູ່ທີ່ອຸນຫະພູມ annealing ປະມານ 5 ℃ສູງກວ່າ Tm ຄາດຄະເນ, ຫຼັງຈາກນັ້ນແຕ່ລະວົງຈອນຫຼຸດລົງ 1 ℃ເຖິງ 2 ℃ຈົນກ່ວາອຸນຫະພູມ annealing ຕ່ໍາກວ່າ Tm 5 ℃.ພຽງແຕ່ແມ່ແບບປາຍທາງທີ່ມີ homology ສູງສຸດທີ່ຈະໄດ້ຮັບການຂະຫຍາຍ.ຜະລິດຕະພັນເຫຼົ່ານີ້ສືບຕໍ່ຂະຫຍາຍຕົວໃນຮອບວຽນຕໍ່ມາ, ການຫຸ້ມຫໍ່ອອກຜະລິດຕະພັນທີ່ບໍ່ແມ່ນການຂະຫຍາຍຕົວ.Descending PCR ແມ່ນເປັນປະໂຫຍດສໍາລັບວິທີການທີ່ລະດັບຄວາມຄ້າຍຄືກັນລະຫວ່າງ primer ແລະ template ເປົ້າຫມາຍແມ່ນບໍ່ຮູ້ຈັກ, ເຊັ່ນ: AFLP DNA fingerprinting.
ຊຸດ PCR ທີ່ກ່ຽວຂ້ອງ
The 2× PCR HeroTMລະບົບປະສົມມີຄວາມທົນທານຕໍ່ PCR inhibitors ສູງກວ່າລະບົບ PCR Mix ທົ່ວໄປ, ແລະສາມາດຮັບມືກັບການຂະຫຍາຍ PCR ຂອງແມ່ແບບສະລັບສັບຊ້ອນຕ່າງໆໄດ້ຢ່າງງ່າຍດາຍ.ລະບົບຕິກິຣິຍາທີ່ເປັນເອກະລັກແລະ Taq Hero ທີ່ມີປະສິດທິພາບສູງເຮັດໃຫ້ປະຕິກິລິຍາ PCR ມີປະສິດທິພາບການຂະຫຍາຍທີ່ສູງຂຶ້ນ, ຄວາມສະເພາະແລະຄວາມອ່ອນໄຫວ.
ປະສິດທິພາບການຂະຫຍາຍທີ່ສູງຂຶ້ນ
ມັນມີກິດຈະກໍາ 5'→3' DNA polymerase ແລະ 5'→3' ກິດຈະກໍາ exonuclease, ໂດຍບໍ່ມີການ 3'→5' ກິດຈະກໍາ exonuclease.
ເວລາຈິງ PCR Easyᵀᴹ-SYBR Green I Kit
ສະເພາະ - buffer ທີ່ດີທີ່ສຸດແລະ enzyme Taq ເລີ່ມຕົ້ນຮ້ອນສາມາດປ້ອງກັນການຂະຫຍາຍທີ່ບໍ່ສະເພາະແລະການສ້າງ primer dimer.
ຄວາມອ່ອນໄຫວສູງ—ສາມາດກວດຫາສຳເນົາຂອງແມ່ແບບຕໍ່າໄດ້
ຊຸດດັ່ງກ່າວໃຊ້ reagent Reverse transcription Foregene ເປັນເອກະລັກແລະ Foregene HotStar Taq DNA Polymerase ປະສົມປະສານກັບລະບົບການຕິກິຣິຍາທີ່ເປັນເອກະລັກເພື່ອປັບປຸງປະສິດທິພາບການຂະຫຍາຍແລະຄວາມຈໍາເພາະຂອງຕິກິຣິຍາ.
ເວລາປະກາດ: 09-09-2023



