



RT-qPCR ຖືກພັດທະນາຈາກເຕັກໂນໂລຢີ PCR ທໍາມະດາ.ມັນເພີ່ມສານເຄມີ fluorescent (ສີຍ້ອມ fluorescent ຫຼື fluorescent probes) ກັບລະບົບປະຕິກິລິຢາ PCR ແບບດັ້ງເດີມ, ແລະກວດພົບຂະບວນການຂັດແລະຂະຫຍາຍ PCR ໃນເວລາທີ່ແທ້ຈິງຕາມກົນໄກ luminescent ທີ່ແຕກຕ່າງກັນ.ການປ່ຽນແປງສັນຍານ fluorescent ໃນຂະຫນາດກາງຖືກນໍາໃຊ້ເພື່ອຄິດໄລ່ປະລິມານການປ່ຽນແປງຂອງຜະລິດຕະພັນໃນແຕ່ລະວົງຈອນຂອງ PCR.ໃນປັດຈຸບັນ, ວິທີການທົ່ວໄປທີ່ສຸດແມ່ນວິທີການຍ້ອມສີ fluorescent ແລະວິທີການ probe.
ວິທີການຍ້ອມສີ fluorescent:
ບາງສີຍ້ອມ fluorescent, ເຊັ່ນ SYBR Green Ⅰ, PicoGreen, BEBO, ແລະອື່ນໆ, ບໍ່ປ່ອຍແສງດ້ວຍຕົວມັນເອງ, ແຕ່ປ່ອຍ fluorescence ຫຼັງຈາກຜູກມັດກັບຮ່ອງເລັກນ້ອຍຂອງ dsDNA.ດັ່ງນັ້ນ, ໃນຕອນເລີ່ມຕົ້ນຂອງປະຕິກິລິຍາ PCR, ເຄື່ອງບໍ່ສາມາດກວດພົບສັນຍານ fluorescent ໄດ້.ເມື່ອປະຕິກິລິຢາກ້າວໄປສູ່ການຍືດຍາວ (ວິທີສອງຂັ້ນຕອນ) ຫຼືຂັ້ນຕອນການຂະຫຍາຍ (ວິທີການສາມຂັ້ນຕອນ), ສາຍສອງແມ່ນເປີດໃນເວລານີ້, ແລະ DNA polymerase ໃຫມ່ໃນລະຫວ່າງການສັງເຄາະ strand, ໂມເລກຸນ fluorescent ຖືກລວມເຂົ້າກັນຢູ່ໃນຮ່ອງເລັກນ້ອຍ dsDNA ແລະປ່ອຍ fluorescence.ເມື່ອຈໍານວນຂອງວົງຈອນ PCR ເພີ່ມຂຶ້ນ, ສີຍ້ອມຫຼາຍແລະຫຼາຍປະສົມປະສານກັບ dsDNA, ແລະສັນຍານ fluorescent ຍັງໄດ້ຮັບການປັບປຸງຢ່າງຕໍ່ເນື່ອງ.ເອົາ SYBR Green Ⅰ ເປັນຕົວຢ່າງ.
ວິທີການສືບສວນ:
Taqman probe ແມ່ນການສືບສວນ hydrolysis ທີ່ໃຊ້ທົ່ວໄປທີ່ສຸດ.ມີກຸ່ມ fluorescent ຢູ່ໃນຕອນທ້າຍຂອງ 5′ ຂອງ probe, ປົກກະຕິແລ້ວ FAM.probe ຕົວຂອງມັນເອງເປັນລໍາດັບທີ່ສົມບູນກັບ gene ເປົ້າຫມາຍ.ມີກຸ່ມ fluorescent quenching ຢູ່ທີ່ 3′ ໃນຕອນທ້າຍຂອງ fluorophore.ອີງຕາມຫຼັກການຂອງ fluorescence resonance ການໂອນພະລັງງານ (Förster resonance ໂອນພະລັງງານ, FRET), ໃນເວລາທີ່ນັກຂ່າວ fluorescent ກຸ່ມ (ໂມເລກຸນ fluorescent ຜູ້ໃຫ້ທຶນ) ແລະກຸ່ມ fluorescent quenching (ໂມເລກຸນ fluorescent ຍອມຮັບ) ໃນເວລາທີ່ spectrum ຕື່ນເຕັ້ນ overlaps ແລະໄລຍະຫ່າງແມ່ນໃກ້ຊິດຫຼາຍ (7-10nmule ຮັບການຍອມຮັບຂອງ fluorescent the molecule), ໄດ້. ໂມເລກຸນ, ໃນຂະນະທີ່ autofluorescence ອ່ອນແອລົງ.ດັ່ງນັ້ນ, ໃນຕອນເລີ່ມຕົ້ນຂອງປະຕິກິລິຍາ PCR, ໃນເວລາທີ່ probe ແມ່ນບໍ່ເສຍຄ່າແລະ intact ໃນລະບົບ, ກຸ່ມ fluorescent ນັກຂ່າວຈະບໍ່ປ່ອຍ fluorescence.ໃນເວລາທີ່ annealing, primer ແລະ probe ຜູກມັດກັບແມ່ແບບ.ໃນລະຫວ່າງຂັ້ນຕອນການຂະຫຍາຍ, polymerase ສືບຕໍ່ສັງເຄາະຕ່ອງໂສ້ໃຫມ່.DNA polymerase ມີກິດຈະກໍາ 5′-3′ exonuclease.ເມື່ອເຖິງການສືບສວນ, DNA polymerase ຈະ hydrolyze probe ຈາກແມ່ແບບ, ແຍກກຸ່ມ fluorescent ນັກຂ່າວອອກຈາກກຸ່ມ fluorescent quencher, ແລະປ່ອຍສັນຍານ fluorescent.ເນື່ອງຈາກມີການພົວພັນຫນຶ່ງຕໍ່ຫນຶ່ງລະຫວ່າງ probe ແລະແມ່ແບບ, ວິທີການ probe ດີກວ່າວິທີການຍ້ອມສີໃນແງ່ຂອງຄວາມຖືກຕ້ອງແລະຄວາມອ່ອນໄຫວຂອງການທົດສອບ.
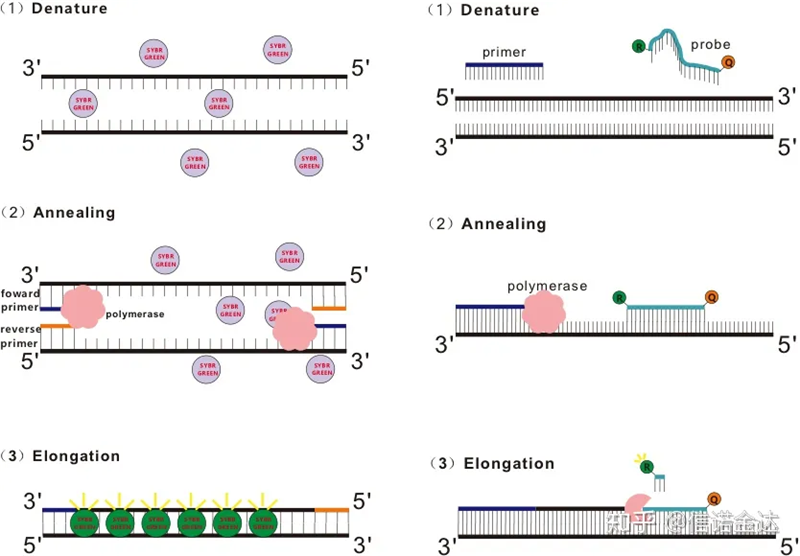
Fig 1 ຫຼັກການຂອງ qRT-PCR
ການອອກແບບ primer
ຫຼັກການ:
primers ຄວນໄດ້ຮັບການອອກແບບຢູ່ໃນເຂດອະນຸລັກຂອງຊຸດອາຊິດນິວຄລີອິກແລະມີຄວາມສະເພາະ.
ມັນດີທີ່ສຸດທີ່ຈະໃຊ້ລໍາດັບ cDNA, ແລະລໍາດັບ mRNA ຍັງເປັນທີ່ຍອມຮັບ.ຖ້າບໍ່ແມ່ນ, ຊອກຫາການອອກແບບພາກພື້ນ cds ຂອງລໍາດັບ DNA.
ຄວາມຍາວຂອງຜະລິດຕະພັນປະລິມານ fluorescent ແມ່ນ 80-150bp, ຍາວທີ່ສຸດແມ່ນ 300bp, ຄວາມຍາວ primer ໂດຍທົ່ວໄປແມ່ນລະຫວ່າງ 17-25 ຖານ, ແລະຄວາມແຕກຕ່າງລະຫວ່າງ primers ເທິງນ້ໍາແລະລຸ່ມນ້ໍາບໍ່ຄວນມີຂະຫນາດໃຫຍ່ເກີນໄປ.
ເນື້ອໃນ G+C ຢູ່ໃນລະຫວ່າງ 40% ຫາ 60%, ແລະ 45-55% ແມ່ນດີທີ່ສຸດ.
ຄ່າ TM ຢູ່ລະຫວ່າງ 58-62 ອົງສາ.
ພະຍາຍາມຫຼີກເວັ້ນການ primer dimers ແລະ self-dimers, (ບໍ່ປາກົດຫຼາຍກ່ວາ 4 ຄູ່ຂອງຖານເສີມຕິດຕໍ່ກັນ) ໂຄງສ້າງ hairpin, ຖ້າຫຼີກລ່ຽງບໍ່ໄດ້, ເຮັດໃຫ້ ΔG<4.5kJ / mol * ຖ້າທ່ານບໍ່ສາມາດຮັບປະກັນວ່າ gDNA ໄດ້ຖືກໂຍກຍ້າຍອອກໃນລະຫວ່າງການຖອດຖອນຄືນໃຫມ່, ມັນດີທີ່ສຸດທີ່ຈະອອກແບບ primers ຂອງ intron * 3′ ຢ່າງຕໍ່ເນື່ອງ, ໂຄງສ້າງຂອງ G / AT / C, ຫຼີກລ້ຽງບໍ່ໄດ້, ໂຄງສ້າງທີ່ອຸດົມສົມບູນ / AT / C. 2-3) primers ແລະ non-
ສະເພາະຄວາມຄ້າຍຄືກັນຂອງລຳດັບທີ່ຂະຫຍາຍຫຼາຍຊະນິດແມ່ນມັກໜ້ອຍກວ່າ 70% ຫຼືມີ 8 ຄວາມຄ້າຍຄືກັນພື້ນຖານ.
ຖານຂໍ້ມູນ:
ຄົ້ນຫາ CottonFGD ໂດຍຄໍາສໍາຄັນ
ການອອກແບບ primer:
ການອອກແບບ primer IDT-qPCR
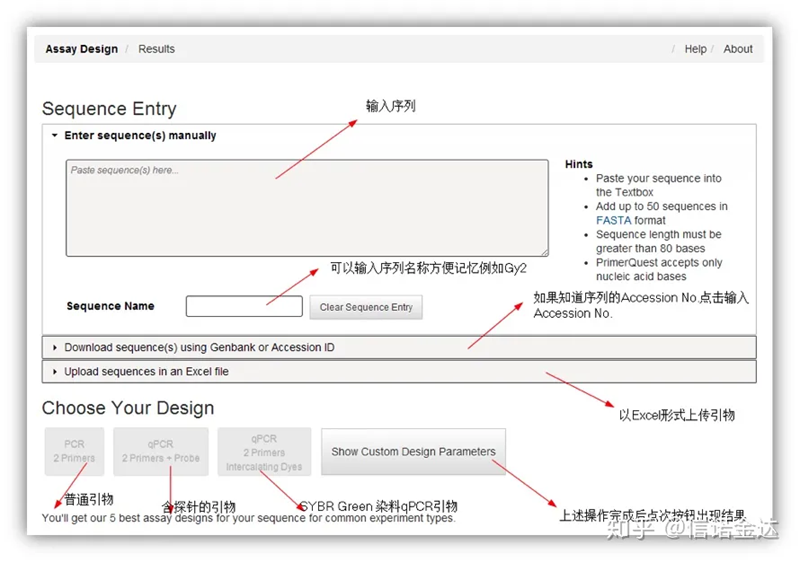
Fig2 IDT ຫນ້າເຄື່ອງມືການອອກແບບ primer ອອນໄລນ໌
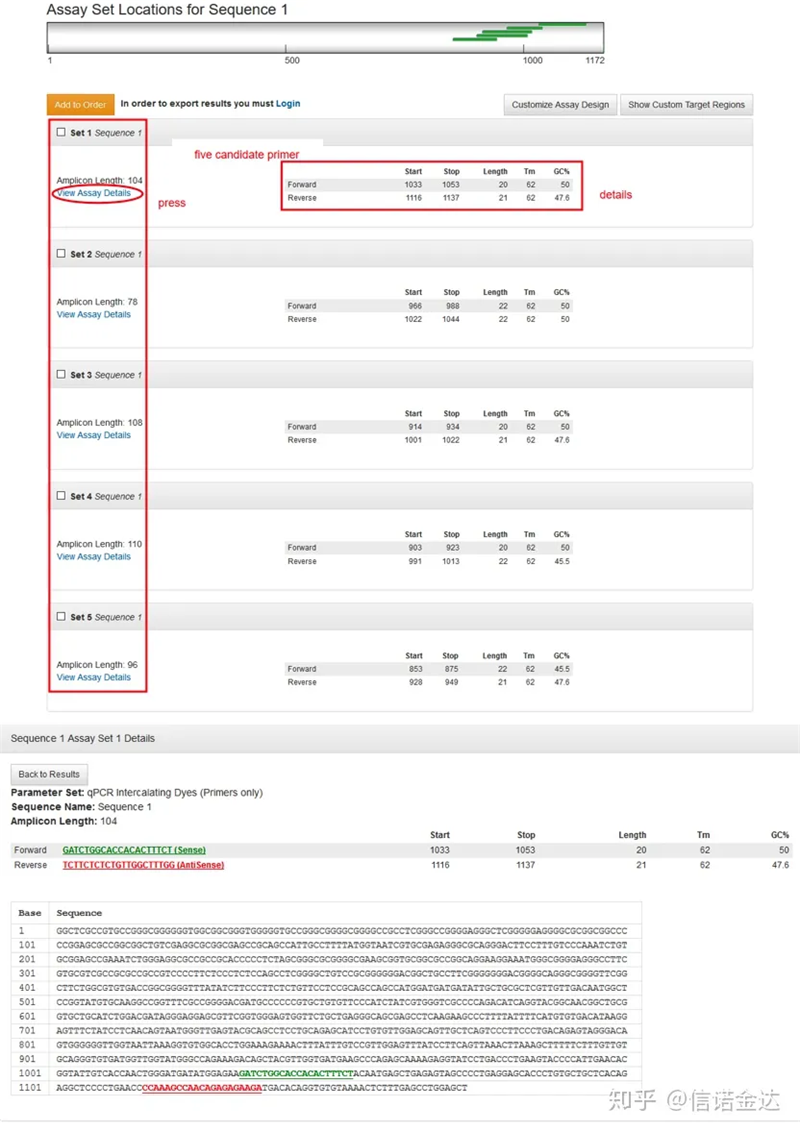
ການສະແດງຜົນຫນ້າ Fig3
ການອອກແບບ primers lncRNA:
lncRNA:ຂັ້ນຕອນດຽວກັນກັບ mRNA.
miRNA:ຫຼັກການຂອງວິທີການ stem-loop: ເນື່ອງຈາກ miRNAs ທັງຫມົດແມ່ນລໍາດັບສັ້ນປະມານ 23 nt, ການກວດສອບ PCR ໂດຍກົງບໍ່ສາມາດປະຕິບັດໄດ້, ດັ່ງນັ້ນເຄື່ອງມືລໍາດັບ stem-loop ຖືກນໍາໃຊ້.ລຳດັບ ລຳ ດັບວົງວຽນແມ່ນ DNA ສາຍດ່ຽວປະມານ 50 nt, ເຊິ່ງສາມາດສ້າງໂຄງສ້າງຂອງ hairpin ດ້ວຍຕົວມັນເອງ.3 'ການສິ້ນສຸດສາມາດອອກແບບເປັນລໍາດັບທີ່ສົມທຽບກັບຊິ້ນສ່ວນຂອງ miRNA, ຫຼັງຈາກນັ້ນ miRNA ເປົ້າຫມາຍສາມາດເຊື່ອມຕໍ່ກັບລໍາດັບລໍາຕົ້ນໃນລະຫວ່າງການຖອດຂໍ້ຄວາມແບບປີ້ນກັບກັນ, ແລະຄວາມຍາວທັງຫມົດສາມາດບັນລຸ 70bp, ເຊິ່ງສອດຄ່ອງກັບຄວາມຍາວຂອງຜະລິດຕະພັນຂະຫຍາຍໃຫຍ່ຂື້ນທີ່ກໍານົດໂດຍ qPCR.ການອອກແບບ primer ຫາງ miRNA.
ການກວດຫາການຂະຫຍາຍສະເພາະ:
ຖານຂໍ້ມູນລະເບີດອອນໄລນ໌: CottonFGD blast ໂດຍຄວາມຄ້າຍຄືກັນລໍາດັບ
ລະເບີດທ້ອງຖິ່ນ: ອ້າງອີງເຖິງການໃຊ້ Blast+ ເພື່ອເຮັດລະເບີດທ້ອງຖິ່ນ, linux ແລະ macos ສາມາດສ້າງຖານຂໍ້ມູນທ້ອງຖິ່ນໂດຍກົງ, ລະບົບ win10 ສາມາດເຮັດໄດ້ຫຼັງຈາກຕິດຕັ້ງ ubuntu bash.ສ້າງຖານຂໍ້ມູນລະເບີດໃນທ້ອງຖິ່ນແລະລະເບີດທ້ອງຖິ່ນ;ເປີດ ubuntu bash ໃນ win10 .
ສັງເກດ: ຝ້າຍເຂດເນີນສູງແລະຝ້າຍເກາະທະເລແມ່ນການປູກພືດ tetraploid, ສະນັ້ນຜົນຂອງການລະເບີດມັກຈະເປັນສອງຫຼືຫຼາຍກ່ວາ.ໃນໄລຍະຜ່ານມາ, ການນໍາໃຊ້ NAU cds ເປັນຖານຂໍ້ມູນເພື່ອປະຕິບັດການລະເບີດມີແນວໂນ້ມທີ່ຈະຊອກຫາສອງ genes homologous ມີພຽງແຕ່ຈໍານວນຫນ້ອຍ SNP ແຕກຕ່າງກັນ.ໂດຍປົກກະຕິແລ້ວ, ສອງພັນທຸ ກຳ ທີ່ເປັນເອກະພາບບໍ່ສາມາດຖືກແຍກອອກໂດຍການອອກແບບ primer, ສະນັ້ນພວກມັນຖືກປະຕິບັດຄືກັນ.ຖ້າຫາກວ່າມີ indel ທີ່ຈະແຈ້ງ, primer ໄດ້ຖືກອອກແບບປົກກະຕິໃນ indel, ແຕ່ນີ້ອາດຈະນໍາໄປສູ່ການໂຄງສ້າງຮອງຂອງ primer ພະລັງງານຟຣີກາຍເປັນທີ່ສູງຂຶ້ນ, ນໍາໄປສູ່ການຫຼຸດລົງຂອງປະສິດທິພາບ amplification, ແຕ່ນີ້ແມ່ນຫຼີກເວັ້ນບໍ່ໄດ້.
ການກວດສອບໂຄງສ້າງຮອງ primer:
ຂັ້ນຕອນ:open oligo 7 → input template sequence → close sub-window → save → locate primer on template, press ctrl+D to set primer length → ວິເຄາະໂຄງສ້າງຮອງຕ່າງໆ, ເຊັ່ນ: ຮ່າງກາຍ dimerization, heterodimer, hairpin, mismatch, ແລະອື່ນໆສອງຮູບສຸດທ້າຍໃນຮູບ 4 ແມ່ນຜົນການທົດສອບຂອງ primers.ຜົນໄດ້ຮັບຂອງ primer ດ້ານຫນ້າແມ່ນດີ, ບໍ່ມີໂຄງສ້າງ dimer ແລະ hairpin ເຫັນໄດ້ຊັດເຈນ, ບໍ່ມີຖານການເສີມຢ່າງຕໍ່ເນື່ອງ, ແລະມູນຄ່າຢ່າງແທ້ຈິງຂອງພະລັງງານຟຣີແມ່ນຫນ້ອຍກວ່າ 4.5, ໃນຂະນະທີ່ primer ດ້ານຫລັງສະແດງໃຫ້ເຫັນຢ່າງຕໍ່ເນື່ອງ 6 ຖານທີ່ປະສົມປະສານ, ແລະພະລັງງານຟຣີແມ່ນ 8.8;ນອກຈາກນັ້ນ, dimer ທີ່ຮຸນແຮງກວ່າຈະປາກົດຢູ່ໃນທ້າຍ 3′, ແລະ dimer ຂອງຖານ 4 ຕິດຕໍ່ກັນປາກົດ.ເຖິງແມ່ນວ່າພະລັງງານຟຣີແມ່ນບໍ່ສູງ, 3′ dimer Chl ສາມາດສົ່ງຜົນກະທົບຢ່າງຮຸນແຮງສະເພາະການຂະຫຍາຍຕົວແລະປະສິດທິພາບການຂະຫຍາຍ.ນອກຈາກນັ້ນ, ມັນຈໍາເປັນຕ້ອງກວດເບິ່ງ hairpins, heterodimers, ແລະບໍ່ກົງກັນ.
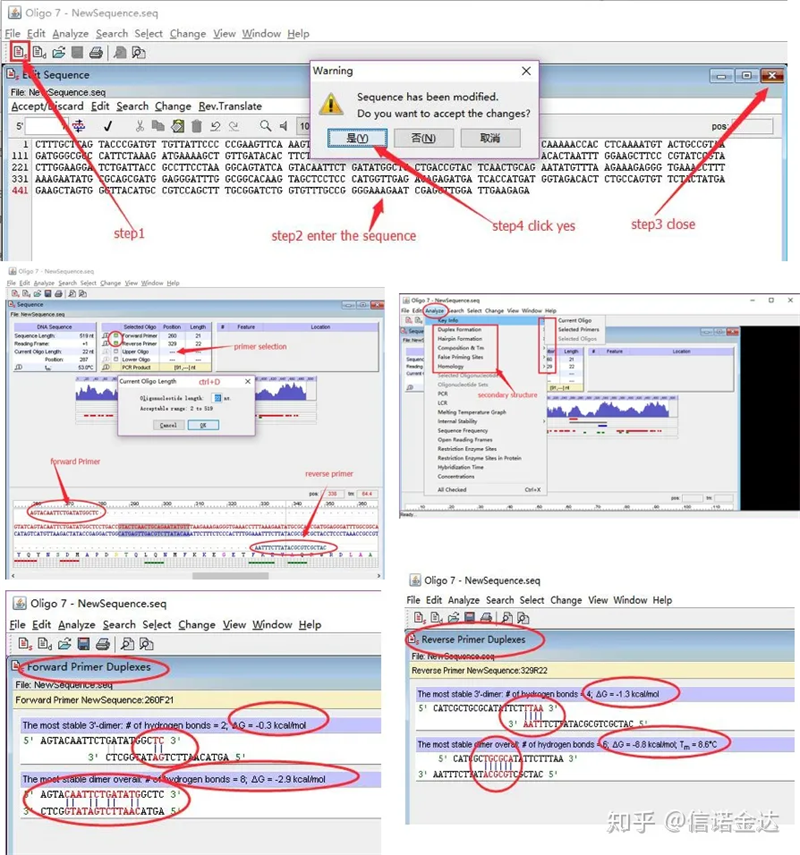
ຜົນການກວດພົບ Fig3 oligo7
ການກວດຫາປະສິດທິພາບການຂະຫຍາຍ:
ປະສິດທິພາບການຂະຫຍາຍຂອງປະຕິກິລິຍາ PCR ມີຜົນກະທົບຢ່າງຮ້າຍແຮງຕໍ່ຜົນຂອງ PCR.ນອກຈາກນີ້ໃນ qRT-PCR, ປະສິດທິພາບການຂະຫຍາຍແມ່ນມີຄວາມສໍາຄັນໂດຍສະເພາະສໍາລັບຜົນໄດ້ຮັບທາງດ້ານປະລິມານ.ເອົາສານອື່ນໆ, ເຄື່ອງຈັກແລະໂປໂຕຄອນໃນ buffer ຕິກິຣິຍາ.ຄຸນນະພາບຂອງ primers ຍັງມີອິດທິພົນຢ່າງຫຼວງຫຼາຍຕໍ່ປະສິດທິພາບການຂະຫຍາຍຂອງ qRT-PCR.ເພື່ອຮັບປະກັນຄວາມຖືກຕ້ອງຂອງຜົນໄດ້ຮັບ, ທັງການກໍານົດປະລິມານ fluorescence ທີ່ກ່ຽວຂ້ອງແລະປະລິມານ fluorescence ຢ່າງແທ້ຈິງຈໍາເປັນຕ້ອງກວດພົບປະສິດທິພາບການຂະຫຍາຍຂອງ primers.ມັນໄດ້ຖືກຮັບຮູ້ວ່າປະສິດທິພາບການຂະຫຍາຍ qRT-PCR ທີ່ມີປະສິດທິພາບແມ່ນຢູ່ລະຫວ່າງ 85% ແລະ 115%.ມີສອງວິທີການ:
1. ວິທີການເສັ້ນໂຄ້ງມາດຕະຖານ:
ກ.ປະສົມ cDNA
ຂ.ການເຈືອຈາງສີ
c.qPCR
ງ.ສົມຜົນ regression ເສັ້ນເພື່ອຄິດໄລ່ປະສິດທິພາບການຂະຫຍາຍ
2. LinRegPCR
LinRegPCR ແມ່ນໂຄງການສໍາລັບການວິເຄາະຂໍ້ມູນ RT-PCR ໃນເວລາທີ່ແທ້ຈິງ, ເຊິ່ງເອີ້ນວ່າຂໍ້ມູນ PCR ປະລິມານ (qPCR) ໂດຍອີງໃສ່ SYBR Green ຫຼືເຄມີທີ່ຄ້າຍຄືກັນ.ໂປລແກລມນໍາໃຊ້ຂໍ້ມູນການແກ້ໄຂທີ່ບໍ່ແມ່ນເສັ້ນພື້ນຖານ, ປະຕິບັດການແກ້ໄຂເສັ້ນພື້ນຖານໃນແຕ່ລະຕົວຢ່າງແຍກຕ່າງຫາກ, ກໍານົດ window-of-linearity ແລະຫຼັງຈາກນັ້ນນໍາໃຊ້ການວິເຄາະການຖົດຖອຍຂອງເສັ້ນກົງເພື່ອໃຫ້ເຫມາະສົມກັບເສັ້ນຊື່ຜ່ານຊຸດຂໍ້ມູນ PCR.ຈາກຄວາມຊັນຂອງເສັ້ນນີ້, ປະສິດທິພາບ PCR ຂອງແຕ່ລະຕົວຢ່າງຖືກຄິດໄລ່.ປະສິດທິພາບ PCR ສະເລ່ຍຕໍ່ amplicon ແລະຄ່າ Ct ຕໍ່ຕົວຢ່າງຖືກນໍາໃຊ້ເພື່ອຄິດໄລ່ຄວາມເຂັ້ມຂົ້ນເລີ່ມຕົ້ນຕໍ່ຕົວຢ່າງ, ສະແດງອອກໃນຫນ່ວຍ fluorescence arbitrary.ການປ້ອນຂໍ້ມູນ ແລະຜົນອອກແມ່ນຜ່ານຕາຕະລາງ Excel.ຕົວຢ່າງເທົ່ານັ້ນ
ການປະສົມແມ່ນຕ້ອງການ, ບໍ່ມີ gradient
ຂັ້ນຕອນທີ່ຕ້ອງການ:(ເອົາ Bole CFX96 ເປັນຕົວຢ່າງ, ບໍ່ແມ່ນເຄື່ອງຈັກທີ່ມີ ABI ທີ່ຊັດເຈນ)
ການທົດລອງ:ມັນເປັນການທົດລອງ qPCR ມາດຕະຖານ.
ຂໍ້ມູນຜົນຜະລິດ qPCR:LinRegPCR ສາມາດຮັບຮູ້ສອງຮູບແບບຂອງໄຟລ໌ຜົນຜະລິດ: RDML ຫຼືຜົນໄດ້ຮັບການຂະຫຍາຍປະລິມານ.ໃນຄວາມເປັນຈິງ, ມັນແມ່ນມູນຄ່າການກວດພົບໃນເວລາທີ່ແທ້ຈິງຂອງຕົວເລກວົງຈອນແລະສັນຍານ fluorescence ໂດຍເຄື່ອງ, ແລະການຂະຫຍາຍແມ່ນໄດ້ຮັບໂດຍການວິເຄາະມູນຄ່າການປ່ຽນແປງ fluorescence ຂອງປະສິດທິພາບສ່ວນເສັ້ນ.
ການເລືອກຂໍ້ມູນ: ໃນທາງທິດສະດີ, ຄ່າ RDML ຄວນສາມາດໃຊ້ໄດ້.ມັນຄາດຄະເນວ່າບັນຫາຂອງຄອມພິວເຕີຂອງຂ້ອຍແມ່ນວ່າຊອບແວບໍ່ສາມາດຮັບຮູ້ RDML, ດັ່ງນັ້ນຂ້ອຍຈຶ່ງມີມູນຄ່າຜົນຜະລິດ excel ເປັນຂໍ້ມູນຕົ້ນສະບັບ.ແນະນໍາໃຫ້ເຮັດການກວດສອບຂໍ້ມູນແບບຫຍາບໆກ່ອນ, ເຊັ່ນຄວາມລົ້ມເຫຼວຂອງການເພີ່ມຕົວຢ່າງ, ແລະອື່ນໆ ຈຸດສາມາດຖືກລຶບຖິ້ມໃນຂໍ້ມູນຜົນຜະລິດ (ແນ່ນອນ, ທ່ານບໍ່ສາມາດລຶບພວກມັນໄດ້, LinRegPCR ຈະບໍ່ສົນໃຈຈຸດເຫຼົ່ານີ້ໃນຂັ້ນຕອນຕໍ່ມາ)
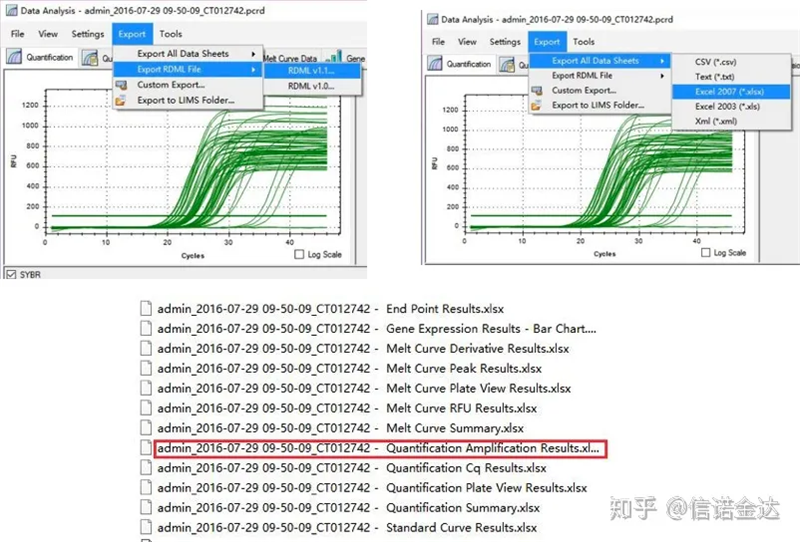
Fig5 qPCR ຂໍ້ມູນສົ່ງອອກ
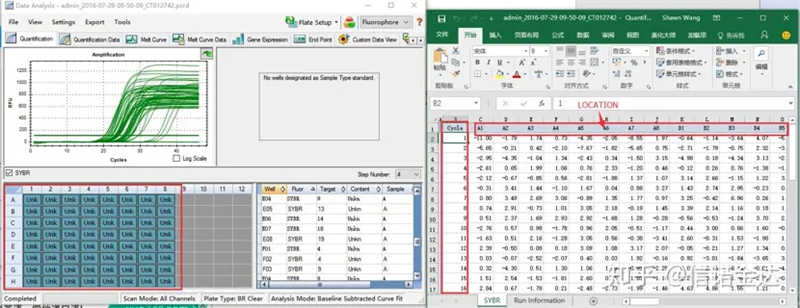
Fig6 ການຄັດເລືອກຕົວຢ່າງຂອງຜູ້ສະຫມັກ
ການປ້ອນຂໍ້ມູນ:ເປີດ qualification amplification results.xls, → ເປີດ LinRegPCR → ໄຟລ໌ → ອ່ານຈາກ excel → ເລືອກພາລາມິເຕີດັ່ງທີ່ສະແດງໃນຮູບ 7 → OK → ຄລິກ ກໍານົດພື້ນຖານ
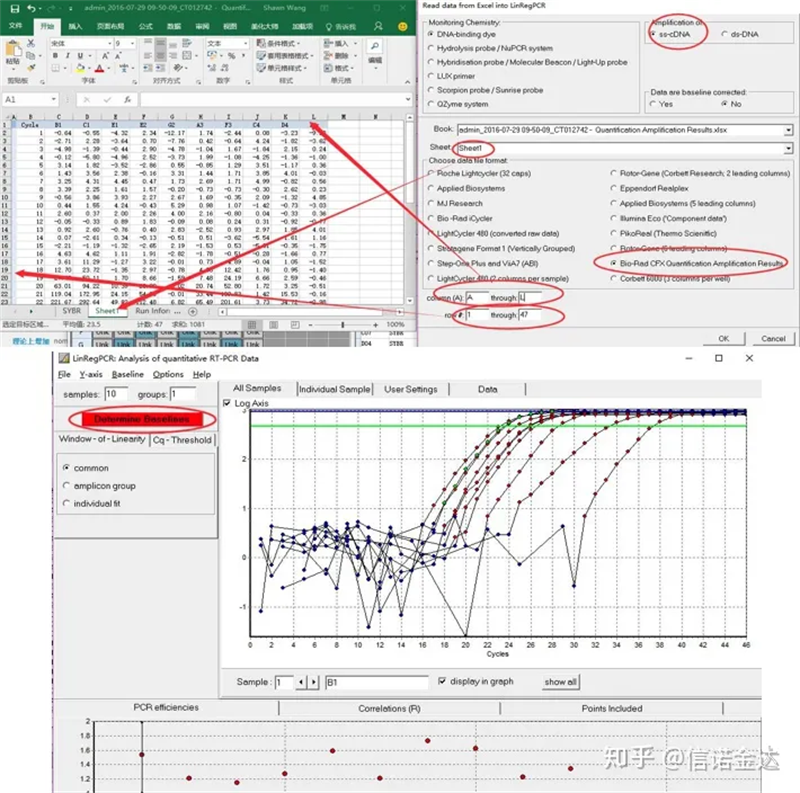
Fig7 ຂັ້ນຕອນຂອງການປ້ອນຂໍ້ມູນ linRegPCR
ຜົນໄດ້ຮັບ:ຖ້າບໍ່ມີການຄ້າງຫ້ອງ, ບໍ່ຕ້ອງຈັດກຸ່ມ.ຖ້າມີການຊໍ້າຊ້ອນ, ການຈັດກຸ່ມສາມາດຖືກດັດແກ້ໃນການຈັດກຸ່ມຕົວຢ່າງ, ແລະຊື່ຂອງ gene ໄດ້ຖືກໃສ່ເຂົ້າໃນຕົວລະບຸ, ແລະຫຼັງຈາກນັ້ນ gene ດຽວກັນຈະຖືກຈັດກຸ່ມໂດຍອັດຕະໂນມັດ.ສຸດທ້າຍ, ໃຫ້ຄລິກໃສ່ໄຟລ໌, ສົ່ງອອກ excel, ແລະເບິ່ງຜົນໄດ້ຮັບ.ປະສິດທິພາບການຂະຫຍາຍ ແລະຜົນ R2 ຂອງແຕ່ລະນ້ຳສ້າງຈະຖືກສະແດງ.ອັນທີສອງ, ຖ້າທ່ານແບ່ງອອກເປັນກຸ່ມ, ປະສິດທິພາບການຂະຫຍາຍສະເລ່ຍທີ່ຖືກແກ້ໄຂຈະຖືກສະແດງ.ໃຫ້ແນ່ໃຈວ່າປະສິດທິພາບການຂະຫຍາຍຂອງແຕ່ລະ primer ແມ່ນຢູ່ລະຫວ່າງ 85% ແລະ 115%.ຖ້າມັນໃຫຍ່ເກີນໄປຫຼືນ້ອຍເກີນໄປ, ມັນຫມາຍຄວາມວ່າປະສິດທິພາບການຂະຫຍາຍຂອງ primer ແມ່ນບໍ່ດີ.
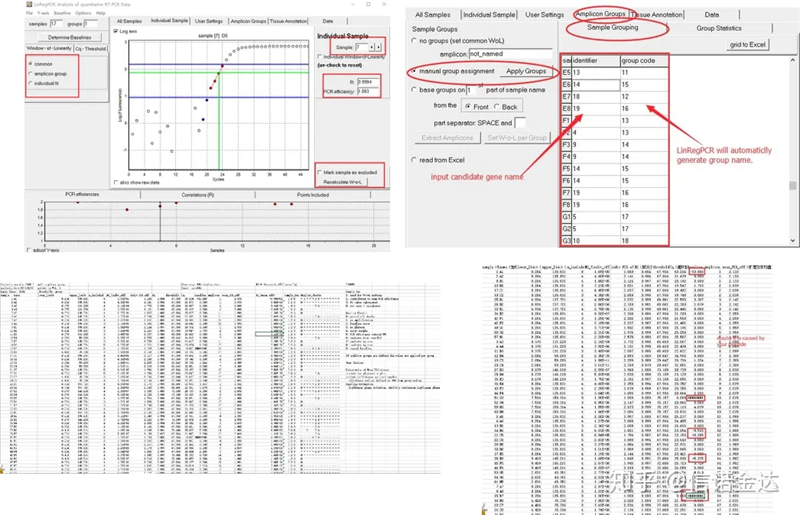
Fig 8 ຜົນໄດ້ຮັບແລະຂໍ້ມູນຜົນຜະລິດ
ຂະບວນການທົດລອງ:
ຄວາມຕ້ອງການຄຸນນະພາບ RNA:
ຄວາມບໍລິສຸດ:1.72.0 ຊີ້ໃຫ້ເຫັນວ່າອາດຈະມີ isothiocyanate ຕົກຄ້າງ.ອາຊິດນິວຄລີອິກສະອາດ A260/A230 ຄວນຈະຢູ່ປະມານ 2 .ຖ້າມີການດູດຊຶມທີ່ເຂັ້ມແຂງຢູ່ທີ່ 230 nm, ມັນຊີ້ໃຫ້ເຫັນວ່າມີທາດປະສົມອິນຊີເຊັ່ນ phenate ions.ນອກຈາກນັ້ນ, ມັນສາມາດກວດພົບໄດ້ໂດຍ 1.5% agarose gel electrophoresis.ຊີ້ໃຫ້ເຫັນເຄື່ອງຫມາຍ, ເພາະວ່າ ssRNA ບໍ່ມີ denaturation ແລະ logarithm ນ້ໍາໂມເລກຸນບໍ່ມີຄວາມສໍາພັນເສັ້ນ, ແລະນ້ໍາໂມເລກຸນບໍ່ສາມາດສະແດງອອກຢ່າງຖືກຕ້ອງ.ຄວາມເຂັ້ມຂຸ້ນ: ທິດສະດີບໍ່ຫນ້ອຍກວ່າ 100ng / ul, ຖ້າຄວາມເຂັ້ມຂົ້ນຕ່ໍາເກີນໄປ, ຄວາມບໍລິສຸດໂດຍທົ່ວໄປແມ່ນຕ່ໍາບໍ່ສູງ
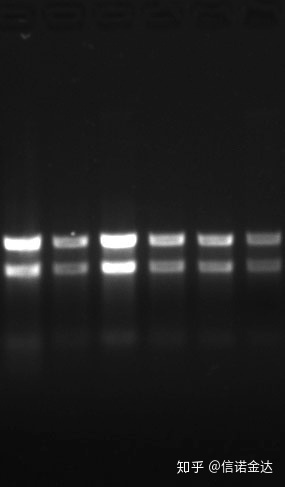
Fig9 RNA gel
ນອກຈາກນັ້ນ, ຖ້າຕົວຢ່າງມີຄ່າແລະຄວາມເຂັ້ມຂົ້ນຂອງ RNA ແມ່ນສູງ, ມັນແນະນໍາໃຫ້ aliquot ມັນຫຼັງຈາກການສະກັດເອົາ, ແລະເຈືອຈາງ RNA ໄປສູ່ຄວາມເຂັ້ມຂົ້ນສຸດທ້າຍຂອງ 100-300ng / ul ສໍາລັບການຖອດຖອນຄືນ.ໃນຂະບວນການຂອງການຖອດຂໍ້ຄວາມແບບປີ້ນກັບກັນ, ເມື່ອ mRNA ຖືກຖອດຂໍ້ຄວາມ, primers oligo (dt) ທີ່ສາມາດຜູກສະເພາະກັບຫາງ polyA ແມ່ນໃຊ້ສໍາລັບການຖອດຂໍ້ຄວາມແບບປີ້ນກັບກັນ, ໃນຂະນະທີ່ lncRNA ແລະ circRNA ໃຊ້ primers ແບບສຸ່ມ hexamer (Random 6 mer) ສໍາລັບການຖອດຂໍ້ຄວາມແບບປີ້ນກັບກັນຂອງ RNA ທັງຫມົດສໍາລັບ miRNA, miRNA-specific miRNA transcription ແມ່ນໃຊ້ສໍາລັບການຖອດຂໍ້ຄວາມແບບສຸ່ມຄໍ.ປະຈຸບັນ, ບໍລິສັດຈໍານວນຫຼາຍໄດ້ເປີດຕົວຊຸດຫາງພິເສດ.ສຳລັບວິທີການໝູນວຽນຕາມລຳຕົ້ນ, ວິທີການຫາງແມ່ນສະດວກກວ່າ, ມີຄວາມສະດວກສູງ, ແລະປະຢັດທາດນ້ຳ, ແຕ່ຜົນກະທົບຂອງການຈຳແນກ miRNAs ຂອງຄອບຄົວດຽວກັນບໍ່ຄວນຈະເປັນຜົນດີເທົ່າກັບວິທີການໝູນວຽນຕາມລຳຕົ້ນ.ແຕ່ລະຊຸດການຖອດຂໍ້ຄວາມແບບປີ້ນກັບກັນມີຄວາມຕ້ອງການສໍາລັບຄວາມເຂັ້ມຂຸ້ນຂອງ primers ສະເພາະພັນທຸກໍາ (stem-loops).ການອ້າງອີງພາຍໃນທີ່ໃຊ້ສໍາລັບ miRNA ແມ່ນ U6.ໃນຂະບວນການປີ້ນກັນຂອງລໍາຕົ້ນ, ທໍ່ຂອງ U6 ຄວນຖືກຖອນຄືນແຍກຕ່າງຫາກ, ແລະ primers ດ້ານຫນ້າແລະດ້ານຫລັງຂອງ U6 ຄວນຖືກເພີ່ມໂດຍກົງ.ທັງ circRNA ແລະ lncRNA ສາມາດໃຊ້ HKGs ເປັນການອ້າງອີງພາຍໃນ.ໃນການກວດສອບ cDNA,
ຖ້າບໍ່ມີບັນຫາກັບ RNA, cDNA ກໍ່ຄວນຈະດີ.ຢ່າງໃດກໍຕາມ, ຖ້າຄວາມສົມບູນແບບຂອງການທົດລອງໄດ້ຖືກປະຕິບັດຕາມ, ມັນດີທີ່ສຸດທີ່ຈະໃຊ້ gene ອ້າງອີງພາຍໃນ (Reference gene, RG) ທີ່ສາມາດຈໍາແນກ gDNA ຈາກ cds.ໂດຍທົ່ວໄປ, RG ແມ່ນ gene ຮັກສາເຮືອນ., HKG) ດັ່ງທີ່ສະແດງຢູ່ໃນຮູບ 10;ໃນເວລານັ້ນ, ຂ້າພະເຈົ້າໄດ້ເຮັດໃຫ້ທາດໂປຼຕີນຈາກ soybean ເກັບຮັກສາໄວ້, ແລະການນໍາໃຊ້ actin7 ທີ່ປະກອບດ້ວຍ introns ເປັນກະສານອ້າງອີງພາຍໃນ.ຂະຫນາດຂອງຊິ້ນສ່ວນຂະຫຍາຍຂອງ primer ນີ້ໃນ gDNA ແມ່ນ 452bp, ແລະຖ້າ cDNA ຖືກໃຊ້ເປັນແມ່ແບບ, ມັນແມ່ນ 142bp.ຫຼັງຈາກນັ້ນ, ຜົນໄດ້ຮັບການທົດສອບພົບວ່າສ່ວນຫນຶ່ງຂອງ cDNA ໄດ້ຖືກປົນເປື້ອນໂດຍ gDNA, ແລະມັນຍັງພິສູດວ່າບໍ່ມີບັນຫາກັບຜົນຂອງການຖອດຂໍ້ຄວາມແບບປີ້ນກັບກັນ, ແລະມັນສາມາດຖືກນໍາໃຊ້ເປັນແມ່ແບບສໍາລັບ PCR.ມັນບໍ່ມີປະໂຫຍດທີ່ຈະດໍາເນີນການ electrophoresis gel agarose ໂດຍກົງກັບ cDNA, ແລະມັນເປັນແຖບກະຈາຍ, ເຊິ່ງບໍ່ຫນ້າເຊື່ອຖື.
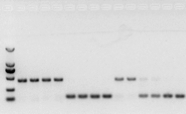
Fig 10 ການກວດຫາ cDNA
ການກໍານົດເງື່ອນໄຂ qPCRໂດຍທົ່ວໄປແລ້ວບໍ່ມີບັນຫາຕາມໂປໂຕຄອນຂອງຊຸດ, ສ່ວນໃຫຍ່ແມ່ນຢູ່ໃນຂັ້ນຕອນຂອງມູນຄ່າ tm.ຖ້າບາງ primers ບໍ່ໄດ້ຮັບການອອກແບບດີໃນລະຫວ່າງການອອກແບບ primer, ເຊິ່ງກໍ່ໃຫ້ເກີດຄວາມແຕກຕ່າງຢ່າງຫຼວງຫຼາຍລະຫວ່າງຄ່າ tm ແລະທິດສະດີ 60 ° C, ມັນແນະນໍາໃຫ້ cDNA ຫຼັງຈາກປະສົມຕົວຢ່າງ, ດໍາເນີນການ PCR gradient ກັບ primers, ແລະພະຍາຍາມຫຼີກເວັ້ນການກໍານົດອຸນຫະພູມໂດຍບໍ່ມີແຖບເປັນຄ່າ TM.
ການວິເຄາະຂໍ້ມູນ
ວິທີການປຸງແຕ່ງ PCR ປະລິມານ fluorescence ປົກກະຕິແມ່ນອີງໃສ່ 2-ΔΔCT.ແມ່ແບບການປະມວນຜົນຂໍ້ມູນ.
ຜະລິດຕະພັນທີ່ກ່ຽວຂ້ອງ:
ເວລາຈິງ PCR ງ່າຍTM -SYBR Green I
RT Easy I (Master Premix ສໍາລັບການສັງເຄາະ cDNA strand ທໍາອິດ)
RT Easy II (Master Premix ສໍາລັບການສັງເຄາະ cDNA strand ທໍາອິດສໍາລັບ qPCR)
ເວລາປະກາດ: 14-03-2023



