



- PCR ແມ່ນວິທີການທີ່ໃຊ້ເພື່ອຂະຫຍາຍ DNA ຈາກຈໍານວນນ້ອຍໆຂອງແມ່ແບບ DNA.RT-PCR ໃຊ້ການຖອດຂໍ້ຄວາມແບບປີ້ນກັບກັນເພື່ອຜະລິດແມ່ແບບ DNA ຈາກແຫຼ່ງ RNA ທີ່ສາມາດຂະຫຍາຍໄດ້.
- PCR ແລະ RT-PCR ປົກກະຕິແລ້ວແມ່ນປະຕິກິລິຍາ endpoint, ໃນຂະນະທີ່ qPCR ແລະ RT-qPCR ໃຊ້ kinetics ຂອງອັດຕາການສັງເຄາະຜະລິດຕະພັນໃນລະຫວ່າງການປະຕິກິລິຍາ PCR ເພື່ອຄິດໄລ່ປະລິມານຂອງແມ່ແບບໃນປະຈຸບັນ.
- ວິທີການໃຫມ່, ເຊັ່ນ PCR ດິຈິຕອນ, ສະຫນອງປະລິມານຢ່າງແທ້ຈິງຂອງແມ່ແບບ DNA ເບື້ອງຕົ້ນ, ໃນຂະນະທີ່ວິທີການເຊັ່ນ: isothermal PCR ຫຼຸດຜ່ອນຄວາມຕ້ອງການອຸປະກອນລາຄາແພງເພື່ອໃຫ້ຜົນໄດ້ຮັບທີ່ເຊື່ອຖືໄດ້.
ປະຕິກິລິຍາລະບົບຕ່ອງໂສ້ Polymerase (PCR) ແມ່ນເຕັກນິກຊີວະວິທະຍາໂມເລກຸນທີ່ຂ້ອນຂ້າງງ່າຍດາຍ ແລະຖືກນໍາໃຊ້ຢ່າງກວ້າງຂວາງເພື່ອຂະຫຍາຍ ແລະກວດຫາລໍາດັບ DNA ແລະ RNA.ເມື່ອປຽບທຽບກັບວິທີການແບບດັ້ງເດີມຂອງ DNA cloning ແລະການຂະຫຍາຍ, ເຊິ່ງມັກຈະໃຊ້ເວລາຫຼາຍມື້, PCR ຕ້ອງການພຽງແຕ່ສອງສາມຊົ່ວໂມງ.PCR ແມ່ນມີຄວາມອ່ອນໄຫວສູງແລະຕ້ອງການແມ່ແບບຫນ້ອຍທີ່ສຸດສໍາລັບການກວດພົບແລະການຂະຫຍາຍຂອງລໍາດັບສະເພາະ.ວິທີການ PCR ພື້ນຖານໄດ້ກ້າວຫນ້າຕື່ມອີກຈາກການກວດພົບ DNA ແລະ RNA ງ່າຍດາຍ.ຂ້າງລຸ່ມນີ້, ພວກເຮົາໄດ້ສະຫນອງພາບລວມຂອງວິທີການ PCR ທີ່ແຕກຕ່າງກັນແລະ reagents ທີ່ພວກເຮົາສະຫນອງໃຫ້ຢູ່ Enzo Life Sciences ສໍາລັບຄວາມຕ້ອງການຄົ້ນຄ້ວາຂອງທ່ານ.ພວກເຮົາມີຈຸດປະສົງເພື່ອຊ່ວຍໃຫ້ນັກວິທະຍາສາດເຂົ້າເຖິງສານ PCR ໄດ້ໄວເພື່ອໃຊ້ໃນໂຄງການຄົ້ນຄ້ວາຕໍ່ໄປຂອງພວກເຂົາ!
PCR
ສໍາລັບ PCR ມາດຕະຖານ, ທັງຫມົດທີ່ທ່ານຕ້ອງການແມ່ນ DNA polymerase, magnesium, nucleotides, primers, ແບບ DNA ທີ່ຈະຂະຫຍາຍ, ແລະ thermocycler.ກົນໄກ PCR ແມ່ນງ່າຍດາຍຄືກັບຈຸດປະສົງຂອງມັນ: 1) DNA ເສັ້ນຄູ່ (dsDNA) ແມ່ນ denatured ຄວາມຮ້ອນ, 2) primers ສອດຄ້ອງກັບສາຍ DNA ດຽວ, ແລະ 3) primers ຖືກຂະຫຍາຍອອກໂດຍ DNA polymerase, ສົ່ງຜົນໃຫ້ສອງສໍາເນົາ. ສາຍ DNA ຕົ້ນສະບັບ.ຂະບວນການ denaturation, annealing, ແລະ elongation ໃນໄລຍະຫຼາຍໆອຸນຫະພູມແລະເວລາແມ່ນເປັນທີ່ຮູ້ຈັກເປັນວົງຈອນຂອງການຂະຫຍາຍ (ຮູບ 1).
|
|
| ຮູບທີ 1.ການສະແດງຕາຕະລາງຂອງວົງຈອນການຂະຫຍາຍໂດຍ PCR. |
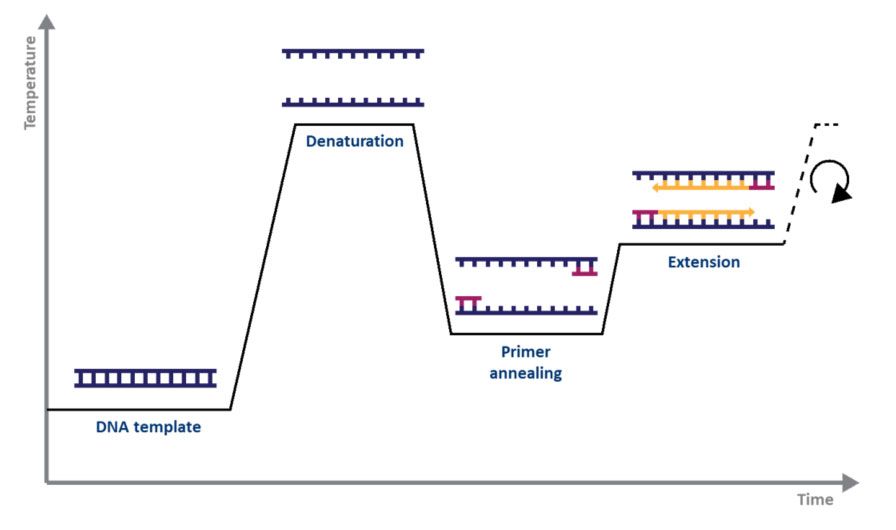
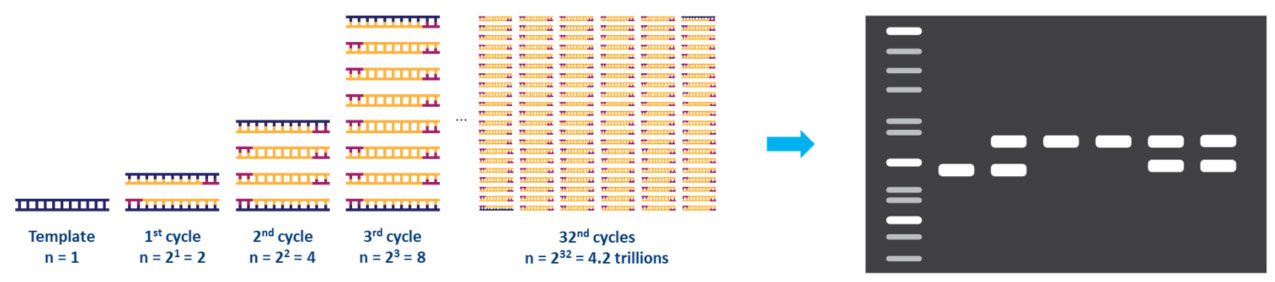
ແຕ່ລະຂັ້ນຕອນຂອງວົງຈອນຄວນຈະຖືກປັບໃຫ້ເຫມາະສົມສໍາລັບຊຸດແມ່ແບບແລະ primer ທີ່ໃຊ້.ວົງຈອນນີ້ຖືກເຮັດຊ້ໍາອີກປະມານ 20-40 ເທື່ອ, ແລະຜະລິດຕະພັນຂະຫຍາຍໃຫຍ່ຂື້ນສາມາດວິເຄາະໄດ້, ໂດຍປົກກະຕິໂດຍ gel agarose (ຮູບ 2).
| |
| ຮູບທີ 2.ການຂະຫຍາຍຕົວແບບ DNA ໂດຍ PCR ແລະການວິເຄາະໂດຍ electrophoresis gel agarose. |
ເນື່ອງຈາກ PCR ເປັນວິທີການທີ່ມີຄວາມອ່ອນໄຫວສູງແລະປະລິມານຂະຫນາດນ້ອຍຫຼາຍແມ່ນຕ້ອງການສໍາລັບປະຕິກິລິຍາດຽວ, ການກະກຽມການປະສົມຕົ້ນສະບັບສໍາລັບປະຕິກິລິຍາຫຼາຍແມ່ນແນະນໍາໃຫ້.ການປະສົມແມ່ບົດຕ້ອງຖືກປະສົມເຂົ້າກັນດີແລະຫຼັງຈາກນັ້ນແບ່ງອອກໂດຍຈໍານວນຂອງຕິກິຣິຍາ, ໃຫ້ແນ່ໃຈວ່າແຕ່ລະຕິກິຣິຍາຈະມີຈໍານວນດຽວກັນຂອງ enzyme, dNTPs, ແລະ primers.ຜູ້ສະຫນອງຈໍານວນຫຼາຍ, ເຊັ່ນ Enzo Life Sciences, ຍັງສະເຫນີການປະສົມ PCR ທີ່ມີທຸກສິ່ງທຸກຢ່າງແລ້ວຍົກເວັ້ນ primers ແລະແມ່ແບບ DNA.
ພາກພື້ນທີ່ອຸດົມສົມບູນ Guanine/Cytosine (GC-rich) ເປັນຕົວແທນຂອງສິ່ງທ້າທາຍໃນເຕັກນິກ PCR ມາດຕະຖານ.ລໍາດັບ GC ທີ່ອຸດົມສົມບູນແມ່ນມີຄວາມຫມັ້ນຄົງຫຼາຍກ່ວາລໍາດັບທີ່ມີເນື້ອໃນ GC ຕ່ໍາ.ນອກຈາກນັ້ນ, ລໍາດັບທີ່ອຸດົມສົມບູນ GC ມີແນວໂນ້ມທີ່ຈະປະກອບເປັນໂຄງສ້າງຂັ້ນສອງ, ເຊັ່ນ: hairpin loops.ດັ່ງນັ້ນ, GC ທີ່ອຸດົມສົມບູນ strands ແມ່ນຍາກທີ່ຈະແຍກອອກຢ່າງສົມບູນໃນໄລຍະການ denaturation.ດັ່ງນັ້ນ, DNA polymerase ບໍ່ສາມາດສັງເຄາະ strand ໃຫມ່ໂດຍບໍ່ມີການຂັດຂວາງ.ອຸນຫະພູມ denaturation ສູງຂຶ້ນສາມາດປັບປຸງສິ່ງນີ້ໄດ້, ແລະການປັບຕົວໄປສູ່ອຸນຫະພູມການຫມູນວຽນທີ່ສູງຂຶ້ນແລະເວລາການຫມຸນທີ່ສັ້ນກວ່າສາມາດປ້ອງກັນການຜູກມັດທີ່ບໍ່ສະເພາະຂອງ primers ທີ່ອຸດົມສົມບູນ GC.reagents ເພີ່ມເຕີມສາມາດເສີມຂະຫຍາຍການຂະຫຍາຍຂອງລໍາດັບ GC ທີ່ອຸດົມສົມບູນ.DMSO, glycerol, ແລະ betaine ຊ່ວຍລົບກວນໂຄງສ້າງຂັ້ນສອງທີ່ເກີດຈາກປະຕິສໍາພັນຂອງ GC ແລະດັ່ງນັ້ນຈຶ່ງອໍານວຍຄວາມສະດວກໃນການແຍກສາຍສອງ.
PCR ເລີ່ມຮ້ອນ
ການຂະຫຍາຍທີ່ບໍ່ສະເພາະແມ່ນບັນຫາທີ່ສາມາດເກີດຂຶ້ນໃນລະຫວ່າງການ PCR.DNA polymerases ສ່ວນໃຫຍ່ທີ່ໃຊ້ສໍາລັບ PCR ເຮັດວຽກດີທີ່ສຸດໃນອຸນຫະພູມປະມານ 68 ° C ຫາ 72 ° C.enzyme ສາມາດ, ຢ່າງໃດກໍຕາມ, ຍັງມີການເຄື່ອນໄຫວຢູ່ໃນອຸນຫະພູມຕ່ໍາ, ເຖິງແມ່ນວ່າໃນລະດັບຕ່ໍາ.ໃນອຸນຫະພູມຕ່ໍາກວ່າອຸນຫະພູມ annealing, primers ສາມາດຜູກມັດທີ່ບໍ່ແມ່ນສະເພາະແລະນໍາໄປສູ່ການຂະຫຍາຍທີ່ບໍ່ສະເພາະ, ເຖິງແມ່ນວ່າປະຕິກິລິຢາຖືກສ້າງຕັ້ງຂຶ້ນໃນກ້ອນ.ນີ້ສາມາດປ້ອງກັນໄດ້ໂດຍການໃຊ້ຕົວຍັບຍັ້ງ polymerase ທີ່ແຍກອອກຈາກ DNA polymerase ພຽງແຕ່ເມື່ອອຸນຫະພູມທີ່ແນ່ນອນມາຮອດ, ດັ່ງນັ້ນຄໍາວ່າ PCR ເລີ່ມຕົ້ນຮ້ອນ.ຕົວຍັບຍັ້ງສາມາດເປັນພູມຕ້ານທານທີ່ຜູກມັດ polymerase ແລະ denatures ຢູ່ທີ່ອຸນຫະພູມເບື້ອງຕົ້ນ (95 ° C ໂດຍປົກກະຕິ).
Polymerase ຄວາມຊື່ສັດສູງ
ໃນຂະນະທີ່ DNA polymerases ຂະຫຍາຍຢ່າງຖືກຕ້ອງກັບລໍາດັບແມ່ແບບຕົ້ນສະບັບ, ຄວາມຜິດພາດໃນການຈັບຄູ່ nucleotide ສາມາດເກີດຂື້ນໄດ້.ຄວາມບໍ່ກົງກັນໃນແອັບພລິເຄຊັນເຊັ່ນ: ການໂຄນເຂົ້າສາມາດສົ່ງຜົນໃຫ້ຂໍ້ຄວາມຖອດຂໍ້ຄວາມຖືກຕັດອອກ, ແລະໂປຣຕີນທີ່ແປຜິດ ຫຼືບໍ່ມີການເຄື່ອນໄຫວລົງລຸ່ມ.ເພື່ອຫຼີກເວັ້ນການບໍ່ກົງກັນເຫຼົ່ານີ້, polymerases ທີ່ມີກິດຈະກໍາ "ການອ່ານຫຼັກຖານ" ໄດ້ຖືກລະບຸແລະລວມເຂົ້າໃນຂະບວນການເຮັດວຽກ.ໂພລີເມີເຣສ ພິສູດຢືນຢັນຕົວທຳອິດ, Pfu, ຖືກລະບຸໃນປີ 1991 ໃນ Pyrococcus furiosus.enzyme Pfu ນີ້ມີກິດຈະກໍາ exonuclease 3' ຫາ 5'.ເມື່ອ DNA ຖືກຂະຫຍາຍອອກ, exonuclease ເອົາ nucleotides ທີ່ບໍ່ກົງກັນຢູ່ປາຍ 3' ຂອງສາຍ.ຫຼັງຈາກນັ້ນ, nucleotide ທີ່ຖືກຕ້ອງຈະຖືກທົດແທນ, ແລະການສັງເຄາະ DNA ຍັງສືບຕໍ່.ການກໍານົດລໍາດັບ nucleotide ທີ່ບໍ່ຖືກຕ້ອງແມ່ນອີງໃສ່ການຜູກມັດສໍາລັບ nucleoside triphosphate ທີ່ຖືກຕ້ອງກັບ enzyme, ບ່ອນທີ່ການຜູກມັດທີ່ບໍ່ມີປະສິດທິພາບເຮັດໃຫ້ການສັງເຄາະຊ້າລົງແລະອະນຸຍາດໃຫ້ມີການທົດແທນທີ່ຖືກຕ້ອງ.ກິດຈະກໍາການພິສູດຂອງ Pfu polymerase ເຮັດໃຫ້ມີຄວາມຜິດພາດຫນ້ອຍໃນລໍາດັບສຸດທ້າຍເມື່ອທຽບກັບ Taq DNA polymerase.ໃນຊຸມປີມໍ່ໆມານີ້, enzymes proofreading ອື່ນໆໄດ້ຖືກລະບຸ, ແລະການດັດແກ້ຂອງ enzyme Pfu ຕົ້ນສະບັບໄດ້ຖືກເຮັດເພື່ອຫຼຸດຜ່ອນອັດຕາຄວາມຜິດພາດໃນລະຫວ່າງການຂະຫຍາຍ DNA.
RT-PCR
Reverse transcription PCR, ຫຼື RT-PCR, ອະນຸຍາດໃຫ້ໃຊ້ RNA ເປັນແມ່ແບບ.ຂັ້ນຕອນເພີ່ມເຕີມອະນຸຍາດໃຫ້ກວດພົບແລະການຂະຫຍາຍ RNA.RNA ຖືກຖອດຂໍ້ຄວາມແບບປີ້ນກັບເຂົ້າເປັນ DNA ເສີມ (cDNA), ໂດຍໃຊ້ reverse transcriptase.ຄຸນນະພາບແລະຄວາມບໍລິສຸດຂອງແມ່ແບບ RNA ເປັນສິ່ງຈໍາເປັນສໍາລັບຄວາມສໍາເລັດຂອງ RT-PCR.ຂັ້ນຕອນທໍາອິດຂອງ RT-PCR ແມ່ນການສັງເຄາະ DNA/RNA ປະສົມ.Reverse transcriptase ຍັງມີຟັງຊັນ RNase H, ເຊິ່ງເຮັດໃຫ້ສ່ວນ RNA ຂອງປະສົມ.ຫຼັງຈາກນັ້ນ, ໂມເລກຸນ DNA ສາຍດຽວແມ່ນສໍາເລັດໂດຍກິດຈະກໍາ DNA polymerase ທີ່ຂຶ້ນກັບ DNA ຂອງ transcriptase ປີ້ນກັບ cDNA.ປະສິດທິພາບຂອງຕິກິຣິຍາສາຍທໍາອິດສາມາດສົ່ງຜົນກະທົບຕໍ່ຂະບວນການຂະຫຍາຍ.ຈາກນີ້, ຂັ້ນຕອນ PCR ມາດຕະຖານຖືກນໍາໃຊ້ເພື່ອຂະຫຍາຍ cDNA.ຄວາມເປັນໄປໄດ້ໃນການປ່ຽນ RNA ເຂົ້າໄປໃນ cDNA ໂດຍ RT-PCR ມີຂໍ້ດີຫຼາຍ, ແລະມັນຖືກນໍາໃຊ້ຕົ້ນຕໍສໍາລັບການວິເຄາະການສະແດງອອກຂອງເຊື້ອສາຍ.RNA ແມ່ນສາຍດຽວແລະບໍ່ຄົງທີ່ຫຼາຍ, ເຊິ່ງເຮັດໃຫ້ມັນທ້າທາຍທີ່ຈະເຮັດວຽກກັບ.ໂດຍທົ່ວໄປແລ້ວມັນເຮັດຫນ້າທີ່ເປັນຂັ້ນຕອນທໍາອິດໃນ qPCR, ເຊິ່ງເຮັດໃຫ້ປະລິມານການຖອດຂໍ້ຄວາມ RNA ໃນຕົວຢ່າງທາງຊີວະພາບ.
qPCR ແລະ RT-qPCR
PCR ປະລິມານ (qPCR) ຖືກນໍາໃຊ້ເພື່ອກວດຫາ, ລັກສະນະແລະປະລິມານອາຊິດ nucleic ສໍາລັບຄໍາຮ້ອງສະຫມັກຈໍານວນຫລາຍ.ໃນ RT-qPCR, RNA transcripts ມັກຈະຖືກຄິດໄລ່ໂດຍການສົ່ງຄືນພວກມັນເຂົ້າໄປໃນ cDNA ທໍາອິດ, ດັ່ງທີ່ໄດ້ອະທິບາຍຂ້າງເທິງ, ແລະຫຼັງຈາກນັ້ນ qPCR ແມ່ນດໍາເນີນການຕໍ່ມາ.ເຊັ່ນດຽວກັນກັບ PCR ມາດຕະຖານ, DNA ໄດ້ຖືກຂະຫຍາຍອອກໂດຍສາມຂັ້ນຕອນຊ້ໍາກັນ: denaturation, annealing, ແລະ elongation.ຢ່າງໃດກໍຕາມ, ໃນ qPCR, ການຕິດສະຫຼາກ fluorescent ຊ່ວຍໃຫ້ການເກັບກໍາຂໍ້ມູນໃນຂະນະທີ່ PCR ກ້າວຫນ້າ.ເຕັກນິກນີ້ມີປະໂຫຍດຫຼາຍອັນເນື່ອງມາຈາກລະດັບຂອງວິທີການແລະເຄມີສາດທີ່ມີຢູ່.
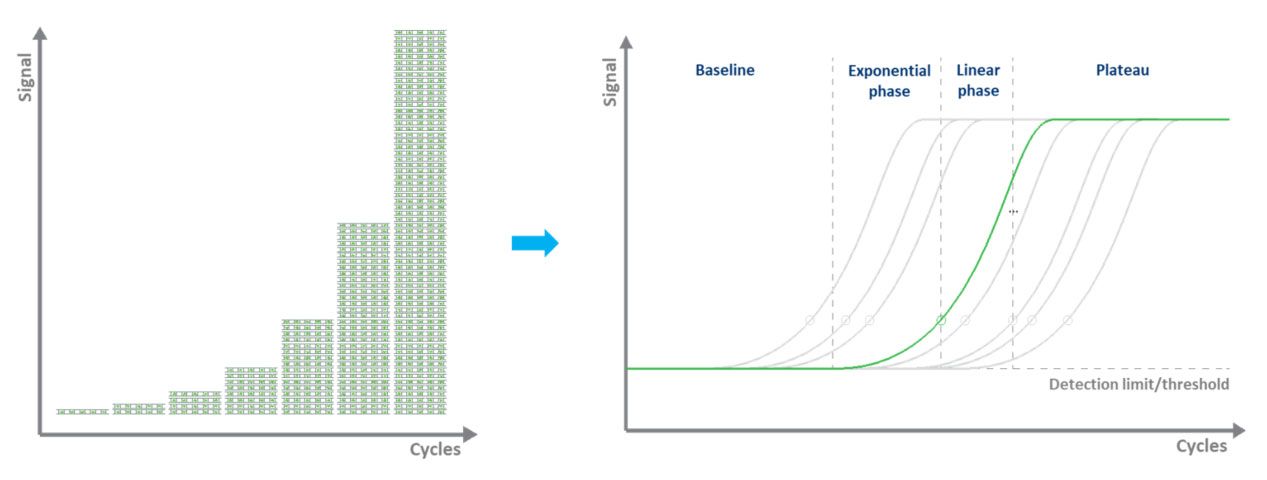
ໃນ qPCR ທີ່ອີງໃສ່ສີຍ້ອມ (ໂດຍປົກກະຕິເປັນສີຂຽວ), ການຕິດສະຫຼາກ fluorescent ອະນຸຍາດໃຫ້ມີປະລິມານຂອງໂມເລກຸນ DNA ທີ່ຂະຫຍາຍໄດ້ໂດຍການໃຊ້ສີຍ້ອມຜ້າທີ່ຜູກມັດ dsDNA.ໃນລະຫວ່າງແຕ່ລະວົງຈອນ, fluorescence ໄດ້ຖືກວັດແທກ.ສັນຍານ fluorescence ເພີ່ມຂຶ້ນຕາມອັດຕາສ່ວນຂອງ DNA ທີ່ຖືກຈໍາລອງ.ດ້ວຍເຫດນີ້, DNA ແມ່ນຖືກຄິດໄລ່ໃນ "ເວລາຈິງ" (ຮູບ 3).ຂໍ້ເສຍຂອງການຍ້ອມສີທີ່ອີງໃສ່ qPCR ແມ່ນມີພຽງແຕ່ຫນຶ່ງເປົ້າຫມາຍທີ່ສາມາດກວດສອບໄດ້ໃນເວລານັ້ນແລະວ່າສີຍ້ອມຈະຜູກມັດກັບ ds-DNA ໃດໆທີ່ມີຢູ່ໃນຕົວຢ່າງ.
|
|
| ຮູບ 3.ການຂະຫຍາຍແມ່ແບບ DNA ໂດຍ qPCR ແລະການວັດແທກສັນຍານ fluorescence ໃນເວລາຈິງ. |
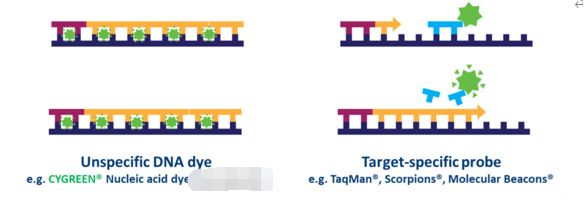
ໃນ qPCR ທີ່ອີງໃສ່ probe, ຫຼາຍເປົ້າຫມາຍສາມາດຖືກກວດພົບພ້ອມໆກັນໃນແຕ່ລະຕົວຢ່າງ, ແຕ່ນີ້ຮຽກຮ້ອງໃຫ້ມີການເພີ່ມປະສິດທິພາບແລະການອອກແບບຂອງ probe ສະເພາະເປົ້າຫມາຍທີ່ໃຊ້ນອກເຫນືອໄປຈາກ primers.ມີຫຼາຍປະເພດຂອງການອອກແບບ probe, ແຕ່ປະເພດທົ່ວໄປທີ່ສຸດແມ່ນ probe hydrolysis, ເຊິ່ງປະກອບດ້ວຍ fluorophore ແລະ quencher.ການຖ່າຍທອດພະລັງງານສະທ້ອນແສງ fluorescence (FRET) ປ້ອງກັນການປ່ອຍອາຍພິດຂອງ fluorophore ຜ່ານ quencher ໃນຂະນະທີ່ probe ແມ່ນ intact.ຢ່າງໃດກໍຕາມ, ໃນລະຫວ່າງການຕິກິຣິຍາ PCR, probe ໄດ້ຖືກ hydrolyzed ໃນລະຫວ່າງການຂະຫຍາຍ primer ແລະການຂະຫຍາຍຂອງລໍາດັບສະເພາະທີ່ມັນຜູກມັດ.ຮອຍແຕກຂອງ probe ແຍກ fluorophore ອອກຈາກ quencher ແລະສົ່ງຜົນໃຫ້ການຂະຫຍາຍ - ຂຶ້ນກັບການເພີ່ມຂື້ນຂອງ fluorescence (ຮູບ 4).ດັ່ງນັ້ນ, ສັນຍານ fluorescence ຈາກປະຕິກິລິຍາ qPCR ທີ່ອີງໃສ່ probe ແມ່ນອັດຕາສ່ວນກັບປະລິມານຂອງລໍາດັບເປົ້າຫມາຍ probe ທີ່ມີຢູ່ໃນຕົວຢ່າງ.ເນື່ອງຈາກວ່າ qPCR ທີ່ອີງໃສ່ probe ແມ່ນສະເພາະຫຼາຍກ່ວາ qPCR ທີ່ມີສີຍ້ອມ, ມັນມັກຈະເປັນເທກໂນໂລຍີທີ່ໃຊ້ໃນການວິເຄາະວິນິດໄສທີ່ອີງໃສ່ qPCR.
| |
| ຮູບ 4.ຄວາມແຕກຕ່າງລະຫວ່າງການຍ້ອມສີທີ່ອີງໃສ່ qPCR ແລະ probe-based probe. |
Isothermal Amplification
ເຕັກນິກ PCR ທີ່ໄດ້ກ່າວມາຂ້າງເທິງນີ້ຮຽກຮ້ອງໃຫ້ມີອຸປະກອນການ thermocycling ລາຄາແພງເພື່ອເຮັດໃຫ້ອຸນຫະພູມຫ້ອງຂຶ້ນແລະຫຼຸດລົງຢ່າງຖືກຕ້ອງສໍາລັບການ denaturation, annealing, ແລະຂັ້ນຕອນການຂະຫຍາຍ.ເຕັກນິກຈໍານວນຫນຶ່ງໄດ້ຖືກພັດທະນາທີ່ບໍ່ຕ້ອງການອຸປະກອນທີ່ຊັດເຈນດັ່ງກ່າວແລະສາມາດປະຕິບັດໄດ້ໃນອາບນ້ໍາແບບງ່າຍດາຍຫຼືແມ້ກະທັ້ງພາຍໃນຈຸລັງທີ່ມີຄວາມສົນໃຈ.ເຕັກນິກເຫຼົ່ານີ້ເອີ້ນວ່າການຂະຫຍາຍ isothermal ແລະເຮັດວຽກໂດຍອີງໃສ່ exponential, linear, ຫຼື cascade amplification.
ປະເພດທີ່ຮູ້ຈັກດີທີ່ສຸດຂອງການຂະຫຍາຍ isothermal ແມ່ນ loop-mediated isothermal amplification, ຫຼື LAMP.LAMP ໃຊ້ການຂະຫຍາຍໂຕເລກທີ່ 65⁰C ເພື່ອຂະຫຍາຍຕົວແບບ DNA ຫຼື RNA.ເມື່ອປະຕິບັດ LAMP, ສີ່ຫາຫົກ primers ເສີມກັບພາກພື້ນຂອງ DNA ເປົ້າຫມາຍແມ່ນຖືກນໍາໃຊ້ກັບ DNA polymerase ເພື່ອສັງເຄາະ DNA ໃຫມ່.ສອງ primers ເຫຼົ່ານີ້ມີລໍາດັບທີ່ສະຫນັບສະຫນູນທີ່ຮັບຮູ້ລໍາດັບໃນ primers ອື່ນໆແລະຜູກມັດພວກມັນ, ອະນຸຍາດໃຫ້ໂຄງສ້າງ "loop" ປະກອບຢູ່ໃນ DNA ທີ່ສັງເຄາະໃຫມ່ທີ່ຊ່ວຍເຮັດໃຫ້ primer annealing ໃນຮອບຕໍ່ໄປຂອງການຂະຫຍາຍ.ໂຄມໄຟສາມາດເບິ່ງເຫັນໄດ້ໂດຍຫຼາຍວິທີ, ລວມທັງ fluorescence, agarose gel electrophoresis, ຫຼື colorimetry.ຄວາມງ່າຍຂອງການເບິ່ງເຫັນແລະກວດພົບການມີຫຼືບໍ່ມີຂອງຜະລິດຕະພັນໂດຍ colorimetry ແລະການຂາດອຸປະກອນລາຄາແພງທີ່ຕ້ອງການເຮັດໃຫ້ LAMP ເປັນທາງເລືອກທີ່ເຫມາະສົມສໍາລັບການທົດສອບ SARS-CoV-2 ໃນເຂດທີ່ຫ້ອງທົດລອງທາງດ້ານການຊ່ວຍບໍ່ສາມາດໃຊ້ໄດ້, ຫຼືການເກັບຮັກສາແລະການຂົນສົ່ງຕົວຢ່າງ. ບໍ່ເປັນໄປໄດ້, ຫຼືຢູ່ໃນຫ້ອງທົດລອງທີ່ບໍ່ມີອຸປະກອນເຄື່ອງຄວບຄຸມຄວາມຮ້ອນ PCR ໃນເມື່ອກ່ອນ.
ເວລາປະກາດ: ສິງຫາ-19-2023



